
Turner's syndrome (TS) is caused by a partial or total deletion of an X chromosome, occurring in 1:2,000 to 1:5,000 live born females. Hearing loss is one of its major clinical manifestations. However, there are few studies investigating this problem.
ObjectivesTo review the current knowledge regarding the epidemiology, etiology, clinical manifestations and diagnosis of hearing impairment in patients with TS.
MethodsA bibliographic search was performed in the Medline and Lilacs databanks (1980-2012) to identify the main papers associating Turner's syndrome, hearing impairment and its clinical outcomes.
ConclusionsRecurrent otitis media, dysfunction of the Eustachian tube, conductive hearing loss during infancy and sensorineural hearing loss in adolescence are the audiologic disorders more common in ST. The karyotype appears to be important in the hearing loss, with studies demonstrating an increased prevalence in patients with monosomy 45,X or isochromosome 46,i(Xq). Morphologic studies of the cochlea are necessary to help out in the clarifying the etiology of the sensorineural hearing loss.
A síndrome de Turner (ST) é causada por uma deleção total ou parcial de um cromossomo X, ocorrendo em 1:2.000 até 1:5.000 meninas nascidas vivas. A perda auditiva é uma de suas principais manifestações clinicas. Entretanto, existem poucos estudos na literatura descrevendo essa associação.
ObjetivoRever o conhecimento atual sobre a epidemiologia, etiologia, manifestações clínicas e diagnósticas da deficiência auditiva em pacientes com ST.
MétodosA pesquisa bibliográfica, realizada entre 1980-2012, utilizou os bancos de dados Medlinee Lilacs identificando os principais artigos que relataram associação entre ST e deficit auditivo e suas repercussões clínicas.
ConclusõesOtite média de repetição, disfunção das trompas de Eustáquio, perdas auditivas condutivas durante a infância e perdas auditivas sensorioneurais a partir da adolescência são os distúrbios audiológicos mais comuns na ST. O cariótipo parece ter relação com a perda auditiva, com estudos mostrando aumento da prevalência de perda auditiva em pacientes com monossomia 45,X e isocromossomos 46,i(Xq). Estudos morfológicos da cóclea são necessários para ajudar a esclarecer a etiologia da perda auditiva sensorioneural.
Turner's syndrome (TS) is a rare disease with known genotypic and phenotypic variability1 that affects females in a ratio of 1:2,000 to 1:5,000 live births.2–6 This syndrome is caused by a partial or total deletion of sexual chromosome. About half of the subjects have a karyotype 45,X; 20%-30% have mosaicism, and the remainder have structural abnormalities.7–9
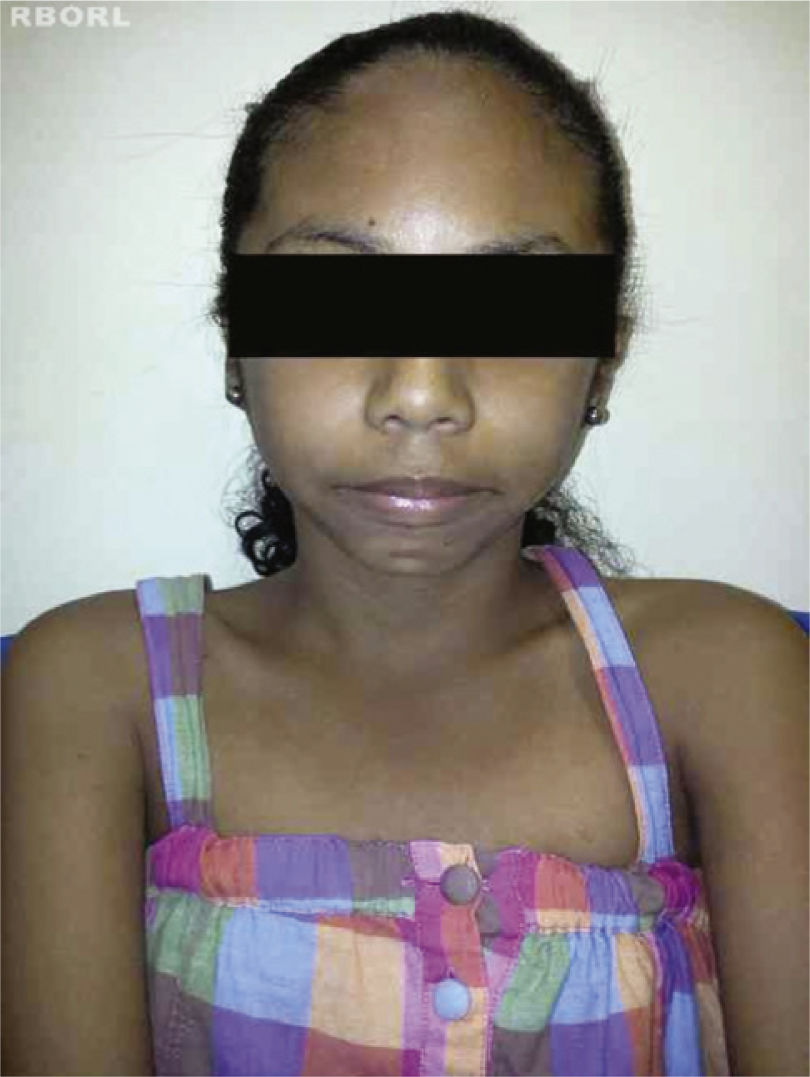
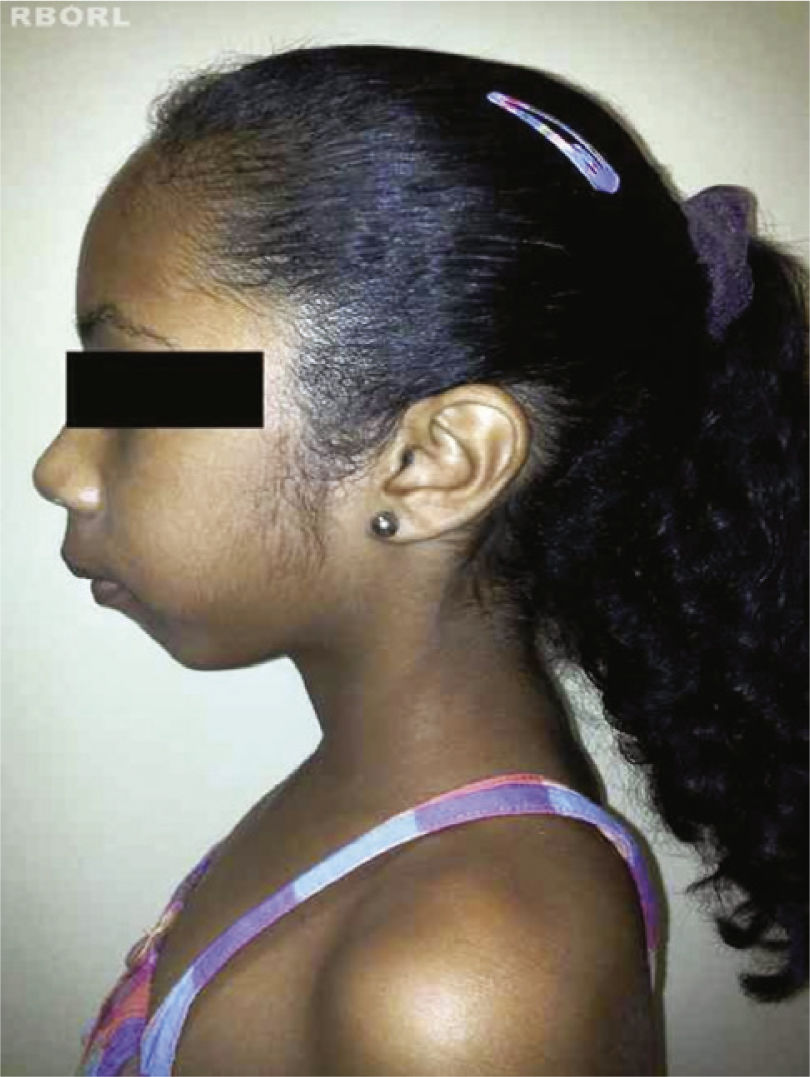
The complete or partial loss of the X chromosome causes the phenotypic manifestations of Turner's syndrome,8 including: short stature, webbing of the neck, low posterior hairline, cubitus valgus, cardiac malformations (e.g., coarctation of the aorta, aortic root dilatation), kidney malformations (e.g., horse shoe kidney), and ovarian dysgenesis2,4,10,11 (Figs. 1 and 2).


The association of otitis media, hearing loss and TS was reported in the early 60's, being confirmed by later studies.12 It is recognized that individuals with TS have a higher incidence of middle ear disease and hearing problems than non-TS subjects.13 The associated hearing impairment has been described as both conductive and sensorineural, indicating both middle and inner ear involvement.13
Recently some studies have described the role of the SHOX (short stature homeobox containing-gene) gene in the pathophysiology of the short stature and hearing problems in TS.14 The short stature is mainly due to SHOX haploinsufficiency and is characterized by a shortfall of approximately 20cm from the predicted adult height.15
The SHOX gene belongs to a family of homeobox genes, transcriptional regulators, and key controllers of the development process. It is expressed within the pharynx involving the first and second arches of the embryo from six weeks of pregnancy on. These arches develop into the maxilla, mandible, and ossicles of the middle ear; muscles involved in opening the Eustachian tube, dampening sounds, chewing, modulating tension of the soft palate, and changing facial expressions; and most of the tongue and outer ear.14 Therefore, in patients with TS, the haploinsufficiency of SHOX expression is a possible explanation for features such as a high palate, prominent ears, chronic otitis media, obstructive sleep apnea, increased sensitivity to noise, and problems such as learning how to suck, blow, eat, and articulate.7,14
Chronic, severe otitis media is common in TS and is thought to be due to obstructed drainage either from lymphatic insufficiency or skeletal dysplasia. The otitis may be associated with a conductive hearing loss in childhood, which usually resolves as otitis decreases in the young adult stage. However, sensorioneural hearing loss affects many young adults with TS and worsens with age. Thus, audiological screening and care are important in both girls and women with TS.15
Among the many chronic health problems faced by individuals with TS, ear disease and hearing loss are two of the most significant and troublesome. Although a number of cross-sectional, mainly retrospective, studies have reported an increased prevalence of otitis media and hearing loss in school-aged girls and women with TS, a no large-scale and prospective study has investigated the prevalence and natural history of ear disease and hearing loss in infants and toddlers.16
In this article, we aimed to review the current knowledge regarding the epidemiology, etiology, clinical manifestations and diagnosis of hearing impairment in patients with TS.
MethodsThe Medline, Lilacs, ProQuest, Cochrane and Embase databases were searched for studies published from 1980-2012. The following keywords, were used combined in different ways: Turner's syndrome; hearing loss; auditory dysfunction; hearing impairment, otitis media, and audiological evaluation. The literature review included consensuses, editorials, original articles and review articles written in English or Portuguese. Studies were initially selected based on their titles and abstracts. The desired outcomes were hearing loss, auditory dysfunction and its clinical manifestations. A review of the bibliographic references of all articles found was performed with the purpose of expanding the search. Sixty articles met the inclusion criteria and, therefore, were searched and included in the present review of literature.
Hearing loss in Turner's syndromeOtitis media is extremely common in girls with TS and about 15% of adults with TS experience hearing loss.17 Hearing loss in Turner's syndrome may be conductive, sensorioneural, or mixed. At least 25% of adults have hearing loss that requires hearing aids. Recurrent otitis media is common, presumably as a result of abnormal Eustachian tube structure and function. Recurrent infections lead to scarring of the tympanic membrane and conductive hearing loss, which may occur even in early childhood. As affected women grow older they become susceptible to a progressive sensorioneural hearing loss that may be related to premature ageing. Because some of these losses may be subtle but functionally impairment, routine hearing evaluation should be carried out through adulthood, particularly in patients with poor school performance.18
Table 1 summarizes the main characteristics of conductive and sensorineural hearing loss in Turner's syndrome.
Main characteristics of Conductive (CHL) and Sensorioneural (SNHL) hearing loss in Turner syndrome.
| Characteristics | CHL | SNHL |
|---|---|---|
| Age | More common in children | More common in adult and elderly |
| Disorders of middle ear | Yes | No |
| Disorders of inner ear | No | Yes |
| Craniofacial malformation | Yes | No |
| Monosomy 45, X | Yes | Yes |
| Recurrent otitis media | Yes | No |
| Decreased serum levels of IGF-1 | Yes | Yes |
| Progressive course | No | Yes |
| Decreased serum levels of estrogens | No | Yes |
| Decreased bone mineral density | Yes | No |
| Inheritance of paternal X | No | Yes |
| Deletion of the short arm ‘p’ of the X chromosome | Yes | Yes |
| Deletion of the long arm ‘q’ of the X chromosome | No | Yes |
| Disturbance of cranial nerve VIII | No | Yes |
| Prevalence | 10% to 47% | 50% to 90% |
| Diagnosis | Audiological evaluation | Audiological evaluation |
| Treatment | Hearing aid | Hearing aid or cochlear implant |
CHL, Conductive Hearing Loss; SNHL, Sensorineural Hearing Loss; IGF-, Insulin-like Growth Factor 1.
Estimatives of the incidence of conductive hearing loss (CHL) in TS are reported to be as high as 80%, with dysfunction of the Eustachian tube and otitis media affecting up to 88% of the patients.19,20 The high prevalence of otitis media in TS is an important factor that possibly influences the individual neuromotor phenotype, and otherwise may cause balance disorders healthy children.21 Besides that, in the context envolving CHL in TS, deafness is common and under-reported.22
The CHL is more prevalent in children and adolescents than in adults, and the frequency of ear infections decreases with aging and growth of facial structures.23 The increased awareness of hearing problems in TS and the correspondent better otologic care may have contributed to a recent decrease in the CHL as a complication of middle ear diseases.24
EtiologyThe main reason for CHL in TS is recurrent persistent otitis media with effusion, chronic middle ear infection, ossicular degeneration following chronic otitis media and cholesteatoma in the middle ear.25 These conditions result from malfunction of the Eustachian tube and anatomic malformation of the skull base, that causes poor drainage and inadequate ventilation of the middle ear and facilitates nasopharyngeal organisms to reach the middle ear.10,25,26 Palatal dysfunction in these patients may be exacerbated by the removal of adenoids.5
Abnormalities of the external ear: low-set and/or cupped auricles, abnormal slopping of the helix downward, forward narrowing of the outer meatus and upward-slanting external auditory canals, are the most common ear anomalies seen in TS.6,27 Besides that, Turner's syndrome is also associated with stapes anomaly and TS patients are thought to have an increased incidence of cleft palate.28 Although computed tomography scanning of the middle ear has not revealed structural anomalies,29 Bergamaschi et al.30 found a significant association between CHL and cranio-facial abnormalities, especially pterygium colli, micrognatia, high arched palate and low-set ears.
The middle ear disorders in TS are, therefore, secondary to abnormalities in the ear anatomy such as: (1) lymphatic hypoplasia causing persistent lymphatic effusion in the middle ear impairing aeration and drainage; (2) abnormal tympanic ostium of the tube; (3) hypotonia of the tensor veli palatine muscle; and (4) brachycephaly an high-arched palate causing abnormally horizontal Eustachian tubes, and possible palatal dysfunction.20,31,32
Apart from inflammation of the middle ear, recent research showed that hearing loss, middle ear infections, and malformations of the external ear in TS are related to the degree of deletion of the p-arm (short arm of the X chromosome) indicating that the hearing loss is also likely associated to the karyotype.26 Some studies have suggested that monosomy 45,X and isochromosome 46,i(Xq) are significantly associated with CHL in TS patients10 indicating that a loss of a gene in the short arm (p) of the chromosome X, like SHOX could be involved.33 Other authors, however, did not find association between CHL and karyotype.30
The otological problems leading to CHL have also being associated with decrease in serum levels of insulin-like growth factor 1 (IGF-1). Barrenäs et al.34 reported that the low levels of IGF-1 correlated with a high occurrence of otitis media. Growth hormone supplementation was effective in ameliorating the otitis media suggesting that this problem could be caused by hypoplasy of the skull base and mastoid cavity.35 In addition, Davenport et al.16 reported evidences that GH treatment does not increase the occurrence of ear problems in young girls with TS. Unfortunately, accurate studies on the effect of growth hormone use and hearing outcomes will be almost impossible, since the majority of TS patients receive growth hormone supplementation.19
The etiology of the recurrent otitis media does not appear to be related to an immunological dysfunction.34
Clinical manifestationsAlthough approximately 30% of 10-years-old girls or younger had a conductive hearing loss,36 as the middle ear infections decrease with age, by late adolescence this complication is almost absent.37 Unlike sensorineural hearing loss, the CHL is not progressive with age.
In addition to the findings above, the incidence of cholesteatoma is higher in TS than in general population, being bilateral in 90% of the cases and diagnosed at the mean age of 15.5 years.30 Women with TS who have low bone mineral density (BMD) and hearing impairment, particularly of the conductive type, are at an increased risk of bone fractures. Improvement of both BMD and hearing ability may help out in reducing fracture risk.38
Sensorineural hearing lossEpidemiologyEar disease in adult patients with TS is complicated by progressive sensorineural hearing loss (SNHL).1 SNHL usually begins in the late childhood-early adulthood in a gradually progressive manner10 and it is a major feature of Turner's syndrome.22 In the specialized literature, incidences of sensorineural hearing loss for populations of patients up to 42 years-old range from 11% to 67%.1 Nonetheless, in one study, 90% of 44% adults with Turner's syndrome had sensorineural hearing loss. The loss was clinically significant in two thirds of the individuals, and 27% of them required hearing aids5. In addition, Kavoussi et al.39 reported that sensorineural hearing loss is extremely common in women with TS, affecting up to 90% of individuals, and consequently causing difficulties at school, work and in social interactions of adolescents.
The incidence of SNHL varies from 16%-60% for mid-frequencies losses to 11%-40% for high-frequencies losses.20 Although the classical audiometric pattern of SNHL in TS is mid-frequency hearing loss, some authors have reported higher incidence of high-frequency pattern.26 The discrepancy among this data may be related to the difference in definition in the dip pattern. Whereas investigators such as Hultcrantz et al.40 define it as an elevation of the threshold exceeding 5dB in the middle frequencies (0.5-2kHz), others, such as Morimoto et al.,26 define it as exceeding 15dB. On the other hand, other investigators define it as exceeding 20dB.25,26 Other possible explanation for the variable results is that the progressive high-frequency hearing loss hides the dip in the middle frequencies.26 Finally, some studies may have included children so young in whom the dip may have not yet developed.25
EtiologyThe causes of high-frequency hearing loss in TS remain controversial. The most frequently studied mechanisms are: age, karyotype (e.g., monosomy vs dissomy, paternal origin of the X chromosome, Xp suppression), estrogen deficiency and/or growth hormone and IGF-1 deficiency and cochlear abnormalities.10,26,37
The SNHL has been attributed to a X-linked dosage effect, because the hearing decline is more common in TS with a complete monosomy for the short arm (p-arm), such as 45,X and 46,Xi(Xq) karyotypes, than in individuals with smaller X deletions.41,42 It is possible that genes, such as SHOX, could induce a delayed cell cycle and fewer sensory cells in the cochlea at birth resulting in cochlear dysfunction.26
The majority of patients with TS inherit their intact X chromosome (Xintact) from their mothers.43 The genomic imprinting, a phenomenon by which there is a differential expression of some genes depending on their chromosome origin have been demonstrated in TS.44 Following this reasoning, Hamelin et al.41 reported that, after pure tone audiometry testing, the prevalence of SNHL was greater in TS patients inheriting the paternal X chromosome (Xpaternal) than Xmaternal subjects, providing evidence of a X-linked effect on the occurrence of SNHL. In addition, TS subjects with a 46,X,i(Xq) karyotype have the highest prevalence of SNHL probably because these individuals inherit more often the Xpaternal chromosome.41,44 These findings suggest that genes expressed in the Xmaternal chromosome may protect against SNHL.41
Abnormal BAEP (Brainstem Auditory Evoked Potentials) in TS suggests impaired cochlear function which may be caused by a neuropathic disturbance or cochlear ganglion defects.13,25 In fact, “Turner mice” have a loss of auditory brainstem response with increased age.40
It has been speculated that estrogens have a protective effect on hearing.45 Wang et al.46 reported that an estrogen receptor-β knockout (BERKO) mouse develops a neural hypocellularity in the somatosensory cortex, and Meltser et al.47 described that the BERKO mice are more susceptible to hearing problems after acoustic problems. Coleman et al.48 reported an improvement of BAEP in ovariectomized rats after estrogen replacement. Ostberg et al.19 reported that the relative risk of developing sensorineural hearing loss in Turner's syndrome was higher if estrogen replacement started above 13 years or if the women were estrogen-deficient for>2 years.
It is not known, however, if the hearing impairment associated with estrogen deficiency is due to poor mineralization of the cochlear capsule or due to the lack of stimulation of estrogen receptors, resulting in an abnormal inner ear development.38 This estrogen protector effect on hearing could not be confirmed by other investigators studying TS patients.19,30
Clinical manifestationsMost commonly, the mid-frequency SNHL in TS begins in the second or third decade (late childhood and early adulthood), although it has been reported in children younger than 10 years.20 With aging, the SNHL develops into a high-frequency hearing loss, resulting, at the age 40, in hearing comparable to that of non-TS women at the age of 60.32,41 It is important, when evaluating TS patients, to rule out common causes of high-frequency hearing loss, such as: ototoxicity and exposure do loud sounds.
Mixed hearing lossMixed hearing loss (MHL) in TS is characterized by the association of middle ear disorders and SNHL. The MHL is diagnosed when the bone conduction thresholds are greater than 15 dBHL and air conduction thresholds greater than 25dB, with air-bone gap greater than or equal to 15, in, at least, one frequency.49 The TS literature is further confused by the inclusion of mixed hearing losses, not separated into conductive and sensorineural components, and often without further identification of the contribution of chronic otitis media to bone-conduction levels.20
Screening for hearing loss in turner syndromeBecause ear disease has a profound impact on the quality of life in TS patients,16 parents and patient (in adolescence) should be made aware of the high prevalence of ear problems at the diagnosis of TS.50 In addition, the audiologist should suspect of TS when evaluating a child with mid-frequency SNHL or chronic otitis media specially if there is an association with short stature, delayed puberty or physical stigmata.25,50
The high incidence of recurrent acute otitis media in girls with TS emphasizes the importance of regular otological check-ups not only to substantiate the diagnosis of patients with TS, but also to monitor these patients for possible conductive or sensorineural hearing loss.8
The following approach is suggested to screen for otological disorders in TS: neonatal screening with BAEP, first audiometry between 18-24 months followed by annual audiometric and otolaringological evaluation. Those with otological problems should be referred for a specialist follow-up.23,30,31
It is important to identify the initial hearing loss at frequencies above 8kHz, before it progress to frequencies critical for verbal communication. When hearing deficits are diagnosed by conventional methods, impairment of communication may have already occurred.31 Thus high frequency audiological testing should be requested for all TS older than 3 years because it can diagnose hearing loss before it is evident at a routine audiometry.25,31 Since the average mid-frequency hearing loss in the Hultcrantz et al.47 study had a mean of 18.8dB, a degree of hearing loss, not detected by the routine 20dB pure-tone hearing screening, is important to obtain air and bone conduction thresholds for any child suspected of having TS.25
Impedance audiometry is indicated to check the condition of the middle ear (e.g., tympanic membrane continuity, middle ear pressure, presence of stapedial muscle reflex); temporal bone computed tomography is indicated for patients with a progressive middle ear disease and cholesteatoma; BAEP should be requested when there is a need to investigate cochlear damage;30 and DPOAEs (Distortion Products Otoacoustic Emission) may be indicated to assess the function of the cochlear microphonics, checking if the outer hearing cells are functioning properly.
ConclusionsOtological disease and hearing loss are frequent in TS. The middle ear disturbances begin in early childhood, and the hearing loss in the first decade. Both can progress to auditory impairment and social disability (e.g., impaired speech or even intellectual development) if they are not diagnosed and treated precociously. The only successful intervention to reduce hearing loss in TS patients is assiduous and appropriate treatment of their auditory problems. Morphologic studies of the organ of Corti and the vestibulocochlear nerve in both fetal and adult TS are necessary to help out in clarifying the etiology of the SNHL.
Conflicts of interestThe authors declare no conflicts of interest.
Please cite this article as: Alves C, Oliveira CS. Hearing loss among with Turner's syndrome: literature review. Braz J Otorhinolaryngol. 2014;80:257-63.


