
Introdução: Mutações no gene da otoferlina (OTOF) são responsáveis pela neuropatia auditiva. Objetivo: Investigar a prevalência de mutações no gene OTOF em pacientes com e sem neuropatia auditiva.
Método: Estudo de casos em corte transversal sendo avaliados 16 casos índice com neuropatia auditiva, 13 pacientes com deficiência auditiva sensorioneural (DASN) e 20 indivíduos ouvintes. DNA foi extraído de leucócitos do sangue periférico e regiões do gene OTOF foram analisadas pela técnica PCR-RFLP.
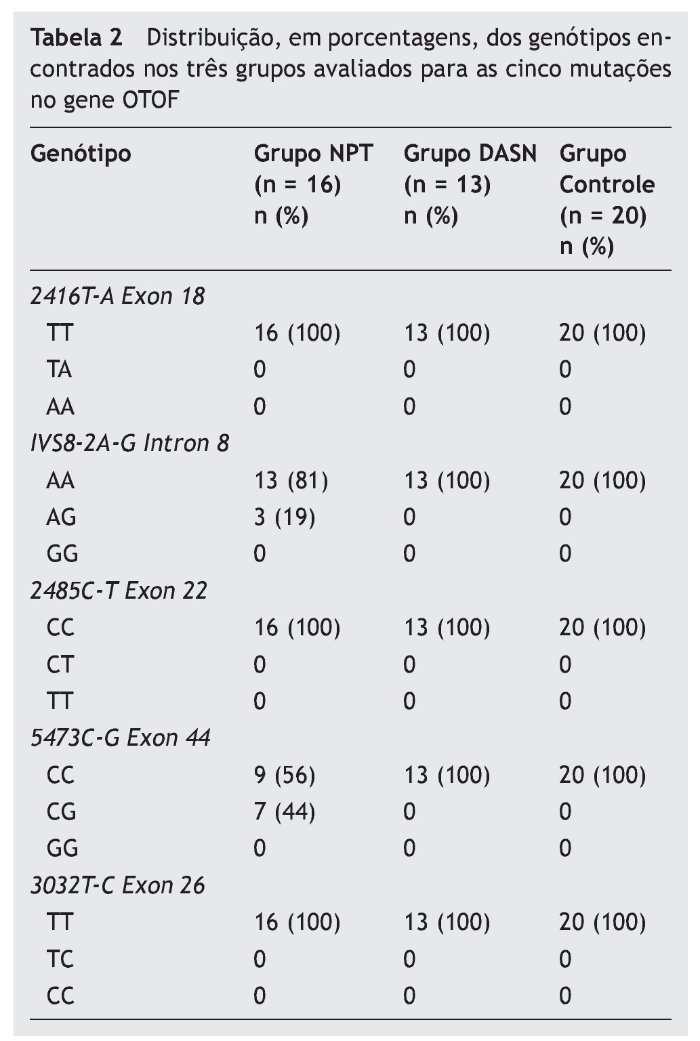
Resultados: Dos 16 casos índice, 9 (56%) são do gênero feminino e 7 (44%) do masculino. Dos 13 pacientes com DASN, 7 (54%) são masculinos e 6 (46%) femininos. Dos 20 ouvintes, 13 (65%) são masculinos e 7 (35%) femininos. Treze (81%) casos índice apresentam o genótipo selvagem (AA) e 3 (19%) o genótipo heterozigoto AG para a mutação IVS8-2A-G (intron 8). A mutação 5473C-G (exon 44) foi encontrada em heterozigose (CG) em 7 (44%) dos casos índice e 9 (56%) apresentam o genótipo selvagem (CC). Destes mutantes, dois (25%) são heterozigotos compostos para as mutações encontradas no intron 8 e exon 44. Os pacientes com DASN e os ouvintes não apresentam mutações (100%).
Conclusão: Existem diferenças, ao nível molecular, em pacientes com e sem neuropatia auditiva.
Introduction: Mutations in the otoferlin gene are responsible for auditory neuropathy. Objective: To investigate the prevalence of mutations in the otoferlin gene in patients with and without auditory neuropathy.
Methods: This original cross-sectional case study evaluated 16 index cases with auditory neuropathy, 13 patients with sensorineural hearing loss, and 20 normal-hearing subjects. DNA was extracted from peripheral blood leukocytes, and the mutations in the otoferlin gene sites were amplified by polymerase chain reaction/restriction fragment length polymorphism.
Results: The 16 index cases included nine (56%) females and seven (44%) males. The 13 deaf patients comprised seven (54%) males and six (46%) females. Among the 20 normal-hearing subjects, 13 (65%) were males and seven were (35%) females. Thirteen (81%) index cases had wild-type genotype (AA) and three (19%) had the heterozygous AG genotype for IVS8-2A-G (intron 8) mutation. The 5473C-G (exon 44) mutation was found in a heterozygous state (CG) in seven (44%) index cases and nine (56%) had the wild-type allele (CC). Of these mutants, two (25%) were compound heterozygotes for the mutations found in intron 8 and exon 44. All patients with sensorineural hearing loss and normal-hearing individuals did not have mutations (100%).
Conclusion: There are differences at the molecular level in patients with and without auditory neuropathy.
Introdução
Depois das mutações no gene GJB2 e GJB6, que são responsáveis por cerca de 80% das deficiências auditivas não sindrômicas autossômicas recessivas, as mutações no gene OTOF (otoferlina) são as mais frequentes das causas genéticas de surdez em crianças. Esse tipo de deficiência auditiva (DA) é mais complicado, uma vez que, na fase inicial, pode apresentar-se como uma neuropatia auditiva (NA), não sendo detectada por triagem auditiva neonatal baseada no teste de emissões otoacústicas.1
A NA é um tipo único de DA quando timpanogramas são normais, os reflexos acústicos e os potenciais evocados auditivos de tronco encefálico (PEATE) são ausentes ou gravemente alterados, mas a função das células ciliadas externas é normal, evidenciada pela presença de emissões otoacústicas (EOAs). Pacientes com essa desordem podem apresentar variações nos graus de DA, além de ter grave prejuízo na compreensão da fala, sendo desproporcional ao grau de perda auditiva.1
Em 2003, foi definido o termo “NA não sindrômica autos-sômica recessiva” devido às características audiológicas e também às descobertas genéticas relacionadas a mutações no gene OTOF que foram associadas a esse tipo de surdez.2 O gene OTOF, localizado no cromossomo 2 (2p23-p22), contém 48 exons e codifica a proteína otoferlina, sendo expresso nas células ciliadas internas cocleares e nas células ciliadas vestibulares Tipo I.3 A partir de então, foram publicados estudos moleculares que permitiram a identificação de mutações no gene OTOF e associálas à NA não sindrômica autossômica recessiva.2-12
Sendo assim, o presente estudo teve como objetivo investigar a prevalência das mutações no gene OTOF: 2416T-A (Y730X) no exon 18,3 IVS8-2-A-G no intron 8/exon 9,6 2485C-T (Q829X) no exon 22,5 5473C-G (Pro1825Ala) no exon 44 5 e 3032T-C (Leu1011Pro) no exon 26,9 em pacientes com NA e em pacientes com DA não sindrômica.
Método
No período de 01/04/2010 a 30/07/2011, foi realizado estudo em corte transversal no qual, dentre 1.230 casos de DA sensorioneural não sindrômica (DASN) do Ambulatório de Surdez da Instituição, foram selecionados 16 pacientes (Grupo NPT – Neuropatia) de ambos os gêneros com NA, os quais foram denominados de casos índice, tendo como critérios de inclusão: exame otoscópico normal; diagnóstico de DASN pela audiometria tonal; presença de EOAs e/ou microfonismo coclear na pesquisa do PEATE; ausência de ondas do PEATE ou alteração grave da morfologia das mesmas; normalidade ao exame de ressonância magnética; ausência dos fatores de risco (infecções maternofetais, meningites, drogas ototóxicas) ou de neuropatias periféricas; e os critérios de exclusão: diagnóstico de perda auditiva condutiva ou mista; presença dos fatores de risco ou de neuropatias periféricas; e idade maior que 65 anos.
Também foram selecionados 13 pacientes (Grupo DASN), de ambos os gêneros, já com diagnóstico de DASN, cujos exames audiológicos não correspondiam ao quadro clínico de NA e sem mutações nos genes da Conexina 26 e GJB6.
Como Grupo Controle (Grupo GC), foram selecionados 20 indivíduos, de ambos os gêneros, sem queixas auditivas, com exame otoscópico normal e sem qualquer parentesco com os pacientes dos Grupos NPT e DASN.
Os casos índice, pacientes e controles eram da mesma origem racial, idade ≤ 65 anos, sem casamento consanguíneo e da mesma área geográfica.
O estudo foi aprovado pelo CEP da Instituição (Parecer n° 44/2010). O DNA genômico foi extraído das amostras de sangue periférico usando-se o kit de extração GE Illustra – Blood Genomicprep Mini Spin Kit™ (GE Healthcare UK Limited), de acordo com o protocolo do fabricante.
Todas as cinco mutações analisadas no gene OTOF foram identificadas pelas suas posições no Gene Bank NCBI RefSeq-Gene: NG_009937.1. Para a análise molecular foram utilizados os testes da PCR/RFLP (Polimerase Chain Reaction/ Restriction Fragment Lenght Polymorphism).
Cada uma das cinco reações da PCR foi processada em ciclador de temperatura (Bioer Technology®, Modelo TC-XPG), em reações de 25 µL de volume final, contendo: 1- DNA (200-300 ng); 2- primers 10 pmoles de cada (direto e inverso); 3- Conjunto de Reagentes FideliTaqTM PCR MasterMix (2×) (GE HEALTHCARE®), com protocolo de acordo com as instruções do fabricante.
Foi utilizado o seguinte ciclo padrão para as reações da PCR, com diferença apenas no tempo de anelamento do primer, que é específico para cada par utilizado: desnaturação inicial a 94 °C por 3 minutos, 35 ciclos repetidos de 60 segundos a 94 °C para desnaturação, 60 segundos a X °C para anelamento do primer (para a mutação 2416T-A: 56 °C; IVS8-2A-G: 51 °C; 2485C-T: 58 °C; mutação 5473C-G: 59 °C; mutação 3032T-C: 52 °C) e extensão de 2 minutos a 72 °C e 10 minutos a 72 °C para extensão final. Os fragmentos amplificados pela PCR foram, respectivamente, submetidos à RFLP, utilizando-se 5U das seguintes enzimas enzima DdeI, BGlII, BfaI, FauI e MspI (New England Biolabs®).
Os produtos das reações (PCR/RFLP) foram adicionados ao azul de bromofenol e submetidos à eletroforese em gel de agarose 2% em tampão Tris-Borato-EDTA 1X contendo brometo de etídio (0,5 µg/mL), tendo sido submetidos à iluminação ultravioleta e os géis fotodocumentados.
Análise estatística
Os resultados foram submetidos previamente à estatística descritiva para determinação da normalidade. Foram utilizados o Teste do Qui-quadrado para comparação entre variáveis independentes com distribuição normal e o Teste de Kruskal-Wallis para amostras com distribuição não normal, além do odds ratio com intervalo de confiança de 95% (95% IC). O nível de significância foi estabelecido em 5%. Testes estatísticos foram realizados usando o programa GraphPad InStat version 3.00 (GraphPad Software Inc., San Diego, Califórnia, EUA). A coerência da distribuição genotípica com o Equilíbrio de Hardy-Weinberg foi avaliada por testes exatos.13
Resultados
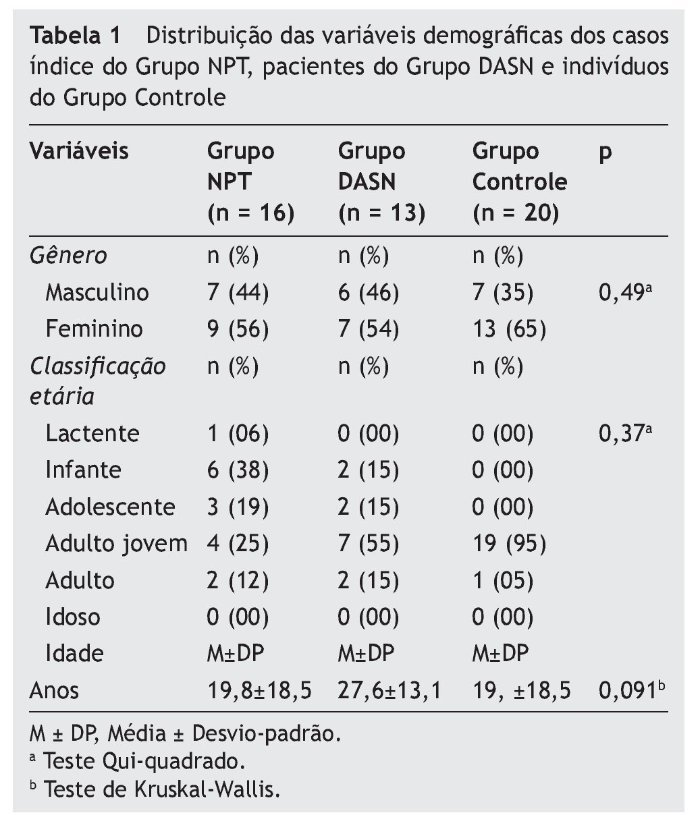
Do total de 16 casos índice do Grupo NPT, nove (56%) eram do gênero feminino e sete (44%) do gênero masculino. Em relação à faixa etária, a idade variou entre oito e 65 anos para o gênero feminino (Média M–24,2 anos; DP ± 20,1), e para o gênero masculino a idade variou entre um e 38 anos (M–14,1 anos; DP ± 15,7), não sendo esta diferença estatisticamente significante (p = 0,29).
Dos 13 pacientes do Grupo DASN, sete (54%) eram do gênero masculino e seis (46%) do gênero feminino. Em relação à faixa etária, a idade variou entre sete e 37 anos para o gênero masculino (M–22,7 anos; DP ± 12,6), e para o gênero feminino a idade variou entre 12 e 45 anos (M–33,3 anos; DP ± 12,3), não havendo significância estatística (p = 0,15).
Em relação aos 20 indivíduos do Grupo Controle, 13 (65%) eram do gênero masculino e sete (35%) do gênero feminino. Em relação à faixa etária, a idade variou entre 19 e 44 anos para o gênero masculino (M–28,5 anos; DP ± 7,2), e para o gênero feminino a idade variou entre 20 e 35 anos (M–26,3 anos; DP ± 6,3), sem diferença estatística significativa (p = 0,49).
Considerando as variáveis demográficas entre os pacientes dos três grupos, houve prevalência do gênero feminino em todos os três, não sendo esta relação estatisticamente significante (p = 0,49). Ambos os grupos, DASN e Controle, tiveram maior prevalência na classificação etária de adulto jovem, perfazendo o total de 55% e 95%, respectivamente. O Grupo NPT teve maior prevalência na faixa infante (38%). A análise estatística para esta variável demográfica não mostrou significância entre os grupos (p = 0,37). Da mesma forma, os valores médios da idade entre os três grupos não foram significantes (p = 0,091) (tabela 1).
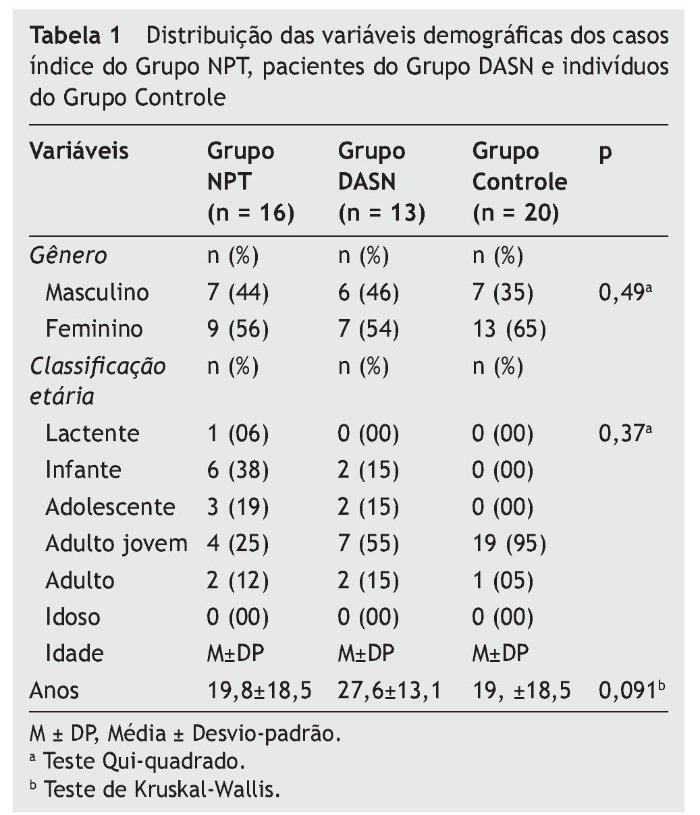
A tabela 2 apresenta a distribuição, em porcentagens, dos genótipos encontrados nos três grupos avaliados para as cinco mutações no gene OTOF. No Grupo NPT, 13 (81%) casos índice apresentaram o genótipo selvagem (AA) e três (19%) o genótipo heterozigoto AG para a mutação IVS8-2A-G (intron 8). A mutação 5473C-G (exon 44) foi encontrada em heterozigose (CG) em sete (44%) dos casos índice e nove (56%) apresentaram o alelo selvagem (CC). Nos Grupos DASN e Controle, 100% dos indivíduos não apresentaram qualquer das mutações analisadas no gene OTOF.
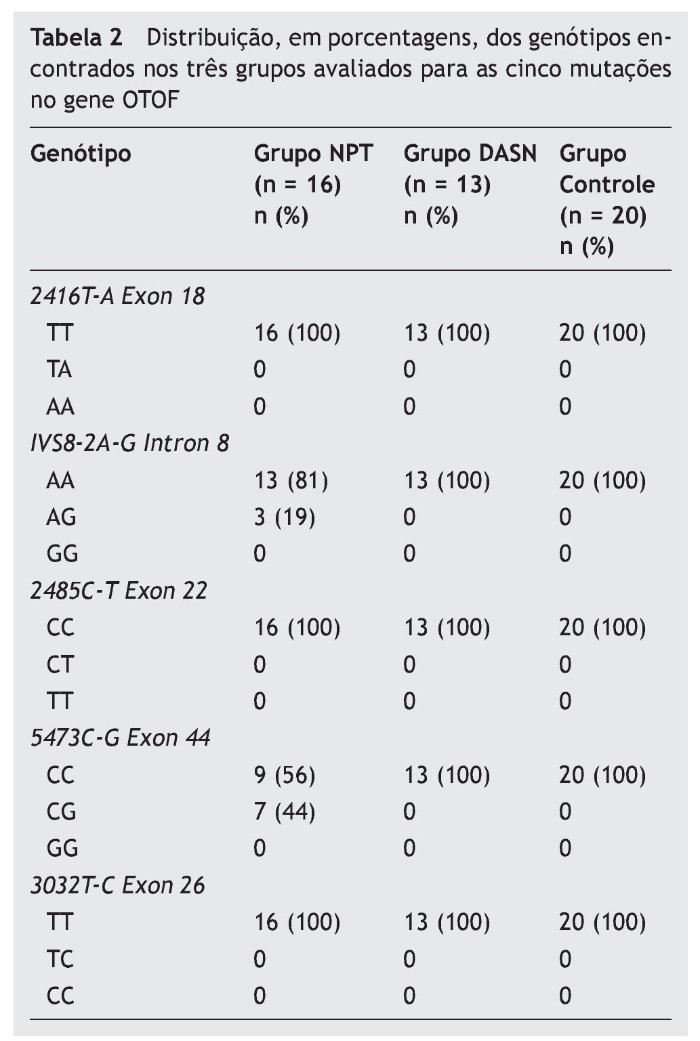
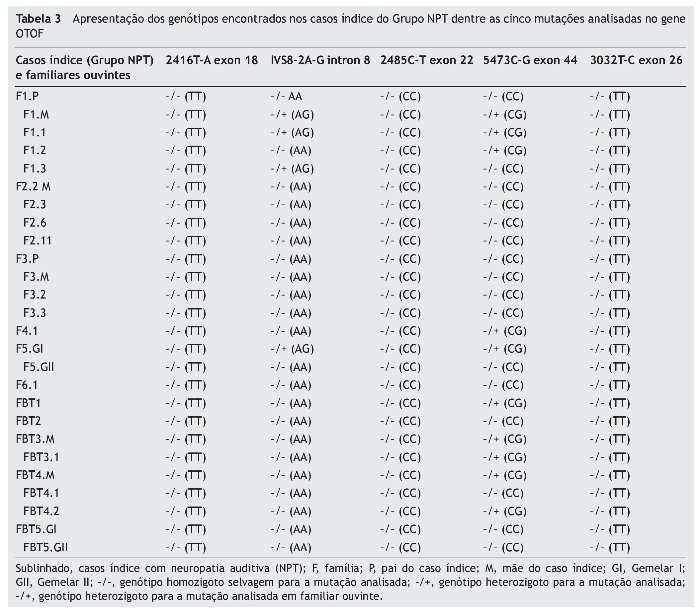
Sendo assim, no Grupo NPT, foram encontrados oito (50%) casos índice com mutações no gene OTOF, sendo um (12,5%; F1.3) heterozigoto para a mutação IVS8-2A-G, cinco (62,5%; F1.2, F4.1, FBT1, FBT3.2 e FBT4.2) heterozigotos para a mutação 5473C-G e dois (25%; F1.1 e F5GI) heterozigotos compostos para as mutações encontradas, respectivamente, no intron 8 e exon 44. Dos familiares ouvintes, a mãe da Família 1 (F1.M) era heterozigota composta para as mutações analisadas no intron 8 e exon 44, e as mães das Famílias FBT3 e FBT4 eram heterozigotas apenas para a mutação 5473C-G (tabela 3).
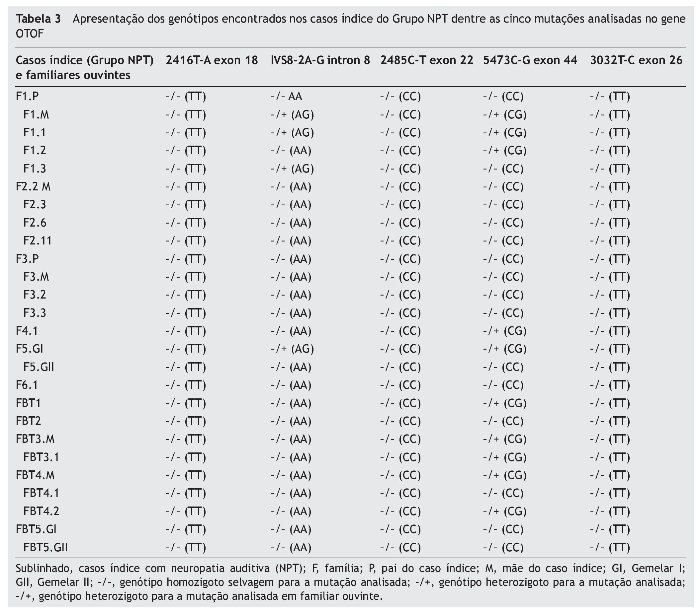
O Grupo NPT foi avaliado para o Equilíbrio de Hardy-Weinberg (EHW) em relação às mutações encontradas no intron 8 e exon 44. De acordo com os cálculos obtidos, as frequências alélicas A (alelo selvagem) e G (alelo mutante) da mutação IVS8-2A-G, para os casos índice do Grupo NPT, foram A = 0,91 e G = 0,09. As frequências genotípicas observadas do homozigoto selvagem (AA-13), heterozigoto mutante (AG-3) e homozigoto mutante (GG-0) destes casos índice se encontravam em concordância com as frequências esperadas pelo EHW (χ2 = 0,171; p = 0,68). Em relação à mutação 5473C-G, as frequências alélicas C (alelo selvagem) e G (alelo mutante), para os casos índice do Grupo NPT, foram C = 0,78 e G = 0,22. Para esta mutação, as frequências genotípicas observadas do homozigoto selvagem (CC-9), heterozigoto mutante (CG-7) e homozigoto mutante (GG-0) destes casos índice também se encontravam em concordância com as frequências esperadas pelo EHW (χ2 = 0,125; p = 0,26).
Em relação ao grau de perda auditiva encontrado nos casos índice do Grupo NPT, três (19%) apresentavam grau leve, cinco (31%) grau moderado, cinco (31%) grau grave e três (19%) grau profundo. No Grupo DASN, quatro (31%) apresentavam grau moderado, dois (15%) grau grave e sete (54%) grau profundo. Não houve casos, neste Grupo, com perda leve. A análise estatística para a variável grau de perda não revelou significância entre ambos os grupos (p = 0,124).
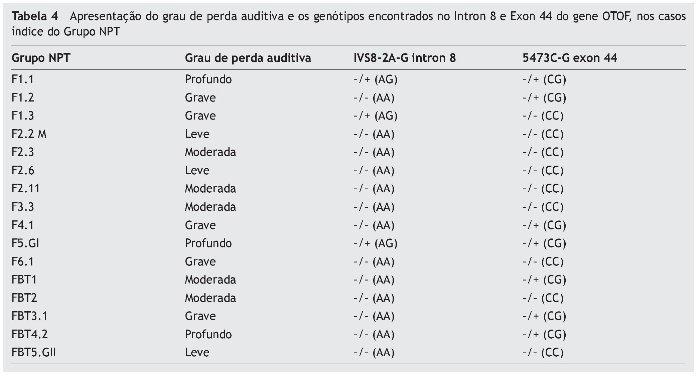
Devido à identificação de mutação somente no intron 8 e exon 44 no gene OTOF, a tabela 4 demonstra os graus de perda auditiva nos casos índice do Grupo NPT e os genótipos encontrados nos mesmos, pela análise molecular dessas regiões.
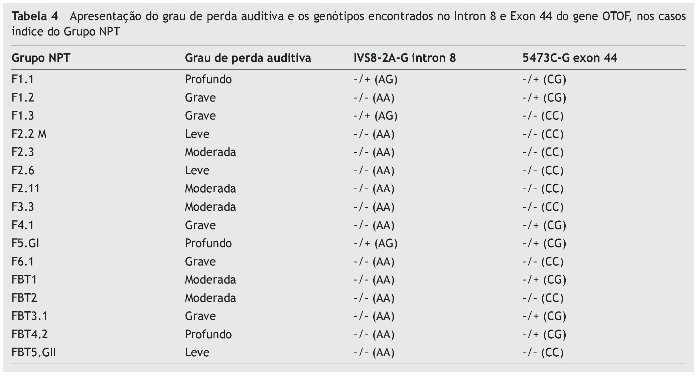
Dentre os casos índice com mutação, os que apresentavam os graus grave (4; 25%) e profundo (3; 19%) foram mais prevalentes do que aqueles sem mutação (1 grau grave apenas, 6%) do mesmo Grupo, os quais apresentaram maior prevalência nos graus leve (3; 19%) e moderado (4; 25%), sendo esta diferença estatisticamente significante (p = 0,022).
Discussão
A NA é um distúrbio auditivo caracterizado pela distorção grave ou ausência do PEATE com as EOAs preservadas, indicando, assim, a presença de integridade das células ciliadas externas.14
Dentre as causas já identificadas de NA, estão a hiperbilirrubinemia, a hipoxia neonatal e/ou doenças neurodegenerativas, mas há relatos de NA “isolada”, isto é, sem qualquer fator de risco associado, não tendo sido proposto qualquer mecanismo fisiopatológico para explicar esses casos. Os recentes avanços na pesquisa genética têm permitido uma nova abordagem para elucidar a fisiopatologia dessa afecção.15,16
Diante do exposto, o presente estudo teve como objetivo investigar a prevalência de cinco mutações no gene OTOF em pacientes com diagnóstico de NA, em indivíduos ouvintes e em pacientes com DASN. Estes foram incluídos a fim de se verificar se mutações no gene OTOF poderiam ser identificadas em ouvintes e em pacientes com DASN sem mutações em outros genes e sem NA.
Em relação às variáveis demográficas analisadas, houve prevalência do gênero feminino nos três grupos do estudo, não sendo esta diferença estatisticamente significante. Os grupos DASN e Controle tiveram maior prevalência na classificação etária de adulto jovem, e o Grupo NPT maior prevalência na faixa infante, dados que não revelaram diferença significativa e que são concordantes com os poucos relatos existentes na literatura sobre a prevalência de gênero e faixa etária relacionados ao gene OTOF.17,18
A maior parte dos relatos de mutações identificadas no gene OTOF tem sido descrita em homozigose e em famílias consanguíneas.3,4,6,9,18-22 Dentre as cinco mutações analisadas no gene OTOF, foram encontradas somente as mutações IVS8-2A-G no intron 8 e a mutação 5473C-G no exon 44, acometendo 50% dos casos índice do estudo. Diferentemente da literatura, todos os casos índice são descendentes de casamentos não consanguíneos, evidenciando que mutações no gene OTOF que estão associadas à NA podem ocorrer nestes casos.
Casos de heterozigose em pacientes com NA foram descritos, associados ou não a casamento consanguíneo.2,5,8,10-12,23,24 No presente estudo, a mutação IVS8-2A-G foi encontrada em heterozigose (um alelo mutante) em 12,5% dos casos índice, o mesmo ocorrendo para a mutação 5473C-G, a qual foi encontrada em 62,5% dos casos. Em 25% dos casos, as duas mutações identificadas ocorreram conjuntamente no mesmo indivíduo, é, por esse motivo, são denominadas heterozigotos compostos. Estas duas mutações, assim como as outras três analisadas, não foram encontradas em 100% do Grupo DASN (100%) e também em 100% do Grupo Controle, dados estes concordantes com as referências.2,5,8,10-12,23,24
Em 2000, a mutação IVS8-2A-G foi primeiramente descrita em homozigose em três irmãos indianos nascidos de casamento consanguíneo, e em heterozigose nos pais e nos irmãos não afetados, não sendo identificada nos 109 indivíduos ouvintes do grupo controle.6 Não há relatos posteriores sobre essa mutação. A mutação 5473C-G foi descrita em heterozigose composta em uma família espanhola, sendo a mutação 2485C-T no exon 22 a identificada no outro alelo.5
Da mesma forma, não há relatos posteriores sobre a referida mutação no exon 44.
A mutação IVS8-2A-G, por criar um prematuro stop codon, e a 5473C-G, por alterar a capacidade de ligação com o Ca+2 no gene selvagem, são consideradas patogênicas, quando em homozigose.3,5 No presente estudo, ambas foram encontradas em heterozigose composta somente nos casos índice com NA (F1.1 e F5.GI), o que poderia explicar a causa molecular da perda auditiva nos mesmos, pois o fenótipo de NA tem sido observado em pacientes portadores de duas mutações afetando todas as isoformas da proteína otoferlina ou uma mutação afetando a isoforma longa e a outra a isoforma curta. Isto significa que qualquer combinação de mutações bialélicas no gene OTOF pode resultar na NA, por serem parcialmente relacionadas à natureza (truncada ou não truncada) ou à localização (isoforma longa ou curta) das mesmas.23
No entanto, na mãe ouvinte (F1.M) do caso índice (F1.1) heterozigoto composto também foram encontradas as mesmas mutações em heterozigose composta. Como a mãe é ouvinte, a causa da DA do caso índice em questão pode ser devida a outra mutação existente em outros exons ou em outros sítios de splice, ou ter mutações mais complexas, tais como grandes deleções ou outros rearranjos de sequência, que necessitariam de abordagem molecular mais específica da que a ora utilizada. Do mesmo modo, estas hipóteses também podem ser aplicadas para explicar a DA nos seis casos índice restantes, por serem portadores de uma das mutações identificadas em apenas um dos alelos.
Referências brasileiras não relataram as mutações identificadas no presente estudo,10-12 sendo que somente um deles identificou a mutação 2485C-T no exon 22 em heterozigose em 0,5% do total de casos de pacientes com DA sensorioneural não sindrômica (1/200).10
As estimativas da prevalência da NA em surdos variam entre 1% a 19%.25-28 No Brasil, a prevalência de NA entre todos os casos de surdez é 1,2%.29 Esta faixa de variação pode se dever ao fato de que diferentes populações foram estudadas ou por diferenças nos critérios utilizados para identificar pacientes com esta afecção. A prevalência de NA dentro de uma população sem fatores de risco não está ainda bem estabelecida.9 No presente estudo todos os critérios de inclusão e exclusão foram rigorosamente seguidos, determinando-se assim a prevalência de 1,3% de casos com NA dentro de uma população com surdez sensorioneural no período estudado.
Também, a estimativa brasileira dos graus de perda nos pacientes com NA varia entre 29,6% grau leve; 55,5% moderado; 7,4% grave e 7,5% profundo.29 No presente estudo, 19% apresentavam grau leve, 31% moderado, 31% grave e 19% profundo. Dentre os casos índice com mutações no gene OTOF, houve prevalência estatisticamente significante dos graus grave e profundo em relação aos graus de perda leve e moderado nos casos índice sem mutação. A intensidade da perda auditiva causada pela NA pode variar de leve a profunda, havendo uma queda desproporcional para a compreensão da palavra falada que não correspondem ao audiograma tonal.
Há certa homogeneidade na descrição do fenótipo dos pacientes com mutações no gene OTOF, o qual é caracterizado por DA congênita ou pré-lingual de grau profundo e não associado a outras anormalidades, conforme os casos índice estudados. Sendo assim, a NA seria a única manifestação fenotípica de mutações no gene OTOF, com distinguível perda auditiva pela relativa preservação da função das células ciliadas externas.9,30
A presença de EOAs nos primeiros meses de vida tem importantes implicações nos programas de rastreamento neonatal, como triagem inicial. Nos casos de NA, existe o risco dessa perda não ser diagnosticada, pois, apesar de estes pacientes terem surdez de grau grave a profundo, contribuiriam para o aumento de casos classificados como “sem alterações ou normais”, pois as EOAs refletem a função auditiva nas células ciliadas externas, e sua presença não exclui alterações no nível das células ciliadas internas, nas sinapses ou no nervo auditivo.28
No contexto da investigação da DA, é importante levar em consideração a existência do tipo de perda da NA, a qual é frequentemente subdiagnosticada. Assim, frente a uma criança ou paciente com surdez e com presença de EOAs, a hipótese de NA tem que ser aventada, e testes genéticos para pesquisa de mutações no gene OTOF devem ser realizados, como executado no presente estudo, permitindo um diagnóstico etiológico preciso a fim de proporcionar tratamento audiológico adequado em tempo hábil.28
As potenciais limitações em relação aos resultados encontrados no estudo merecem consideração. Apesar dos resultados positivos, os mesmos devem ser interpretados com atenção e devem ser corroborados por estudos independentes e multicêntricos para determinar a real prevalência de mutações no gene OTOF e a associação com a NA na população brasileira.
Conclusão
As evidências de associação entre mutações no gene OTOF e a NA permitem concluir que: existem diferenças, em nível molecular, entre pacientes com e sem este tipo de perda auditiva; na ausência fatores de risco, a combinação de distorção grave ou ausência do PEATE e presença das EOAs deve direcionar ao diagnóstico de NA e ao rastreamento de mutações no gene OTOF; e os testes moleculares são valiosos complementos de diagnóstico não apenas individual, mas também como método de rastreamento para estudo populacional ou neonatal.
Financiamento
Este estudo foi financiado pela Bolsa-auxílio Pesquisa (BAP-FAMERP).
Conflitos de interesse
Os autores declaram não haver conflitos de interesse.
Agradecimentos
Aos pacientes e responsáveis que, por concordarem com a execução desta pesquisa, contribuirão para um futuro melhor às crianças de nosso país. À Instituição na qual a pesquisa foi realizada, pelo incentivo à pesquisa e à qualificação docente.
Recebido em 4 de julho de 2014;
aceito em 12 de agosto de 2014
DOI se refere ao artigo: http://dx.doi.org/10.1016/j.bjorl.2015.03.005
☆ Como citar este artigo: da Silva MA, Piatto VB, Maniglia JV. Molecular approach of auditory neuropathy. Braz J Otorhinolaryngol. 2015;81:321-8.
* Autor para correspondência.
E-mails: vbpiatto@gmail.com, vania.piatto@famerp.br (V.B. Piatto).


