
Introdução: Uma etapa fundamental do desenvolvimento do câncer é o acúmulo progressivo de alterações genômicas, resultando na ruptura de vários mecanismos biológicos. Carcinoma ex-adenoma pleomórfico (CXAP) é uma neoplasia agressiva que surge a partir de um adenoma pleomórfico. O CXAP derivado de um AP recorrente (APR) foi raramente relatado e, até o momento, as alterações genômicas associadas a esses tumores não foram estudados.
Objetivo: Avaliar as diferenças entre os CXAPs decorrentes de APRs e os APRs sem transformações malignas usando hibridização genômica comparativa em microarrays (array Comparative Genomic Hibridization – aCGH) a fim de identificar alterações no número de cópias somáticas e os genes afetados.
Método: Amostras de DNA extraídas de tumores provenientes de tecido emblocado em parafina foram submetidos à investigação com a técnica aCGH, e os dados foram analisados com o Nexus Copy Number Discovery Edition v.7.
Resultados: Não observamos alterações no numero de cópias somáticas nos APRs sem transformação maligna. Quanto ao CXAP de APR, embora os perfis genômicos sejam exclusivos para cada caso, detectamos algumas regiões cromossômicas que pareciam ser preferencialmente afetadas por alterações no número de cópias. O primeiro caso de CXAP-APR (carcinoma mioepitelial francamente invasivo) apresentou alterações no numero de cópias afetando 1p36.33p13, 5p e cromossomos 3 e 8. O segundo caso de CXAP-APR (carcinoma epitelialmioepitelial francamente invasivo) apresentou várias alterações nos cromossomos 3, 8 e 16, com duas amplificações em 8p12p11.21 e 12q14.3q21.2. O terceiro caso de CXAP-APR (carcinoma epitelial-mioepitelial minimamente invasivo) apresentou amplificações em 12q13.3q14.1, 12q14.3, e 12q15.
Conclusão: A ocorrência de ganhos de cromossomos 3 e 8, e as amplificações genômicas em 8p e 12q, principalmente aquelas que englobam os HMGA2, MDM2, WIF1, WHSC1L1, RG3, CDK4 no CXAP decorrente de APR podem ser fatores promocionais significativos para a transformação maligna.
Introduction: A key step of cancer development is the progressive accumulation of genomic changes resulting in disruption of several biological mechanisms. Carcinoma ex-pleomorphic adenoma (CXPA) is an aggressive neoplasm that arises from a pleomorphic adenoma. CXPA derived from a recurrent PA (RPA) has been rarely reported, and the genomic changes associated with these tumors have not yet been studied.
Objective: We analyzed CXPA from RPAs and RPAs without malignant transformation using arraycomparative genomic hybridization (array-CGH) to identify somatic copy number alterations and affected genes.
Methods: DNA samples extracted from FFPE tumors were submitted to array-CGH investigation, and data was analyzed by Nexus Copy Number Discovery Edition v.7.
Results: No somatic copy number alterations were found in RPAs without malignant transformation. As for CXPA from RPA, although genomic profiles were unique for each case, we detected some chromosomal regions that appear to be preferentially affected by copy number alterations. The first case of CXPA-RPA (frankly invasive myoepithelial carcinoma) showed copy number alterations affecting 1p36.33p13, 5p and chromosomes 3 and 8. The second case of CXPARPA (frankly invasive epithelial-myoepithelial carcinoma) showed several alterations at chromosomes 3, 8, and 16, with two amplifications at 8p12p11.21 and 12q14.3q21.2. The third case of CXPA-RPA (minimally invasive epithelial-myoepithelial carcinoma) exhibited amplifications at 12q13.3q14.1, 12q14.3, and 12q15.
Conclusion: The occurrence of gains at chromosomes 3 and 8 and genomic amplifications at 8p and 12q, mainly those encompassing the HMGA2, MDM2, WIF1, WHSC1L1, LIRG3, CDK4 in CXAP from RPA can be a significant promotional factor in malignant transformation.
Introdução
O adenoma pleomórfico (AP) é o tumor mais comum das glândulas salivares e representa cerca de 60% a 70% dessas neoplasias. É um tumor benigno com alto risco de recorrência e transformação maligna.1 O risco de recorrência varia de 0,4% a 45%, dependendo da técnica cirúrgica2: 20% a 45% após enucleação, 2% a 5% após lobectomia da parótida e até 0,4% após parotidectomia radical.3 O adenoma pleomórfico recorrente (APR) presumidamente decorre da ruptura de cápsula, ressecção incompleta de extensões microscópicas além da pseudocápsula, ou por origem multifocal.4
Risco de lesão permanente do nervo facial, característica multinodular e aumento da frequência de nova recidiva são fatores que dificultam o tratamento do APR.5 Além disso, o risco de transformação maligna aumenta com o tempo de doença.6,7 Até a presente data, o CXAP decorrente de APR foi raramente relatado e os estudos concentraram-se nas características histopatológicas e clínicas das lesões.8
A recorrência do tumor pode ser causada por aumento no número ou na complexidade das alterações genéticas ou por aquisição de mutações que promovem alterações malignas. O câncer é impulsionado por mutações adquiridas somaticamente, e acredita-se que os rearranjos cromossômicos se acumulam gradualmente ao longo do tempo.9 A triagem de todo o genoma, como a realizada por Hibridação Genômica Comparativa em microarrays (array Comparative Genomic Hybridization – aCGH), pode ser aplicada para revelar alterações no número de cópias que podem identificar a base molecular para a carcinogênese.
Neste estudo, o objetivo foi investigar por meio de aCGH o perfil genômico das alterações no número de cópias associadas a três casos de carcinoma ex-adenoma pleomórfico (CXAP) decorrentes de APR, identificar os genes envolvidos e comparar os resultados aos de quatro casos de APR sem transformação maligna.
Método
O presente estudo foi conduzido de acordo com as normas éticas da nossa instituição (Processo nº CEP/FOP 002/2011). As amostras de DNA foram extraídas de um cilindro com 1,5 mm de diâmetro de tecidos tumorais embebidos em parafina usando o kit de extração Qiagen (Qiagen GmbH, Hilden, Alemanha), de acordo com as instruções do fabricante. O protocolo incluiu desparafinização com xileno, seguida por lavagens com metanol e 24 horas de incubação em 1 mol/L de tiocianato de sódio. Subsequentemente, o sedimento dos tecidos foi seco e digerido por 1,5 dias em tampão de lise com nível alto de proteinase K (60 uL). As amostras foram purificadas em coluna antes da eluição com tampão.
Amostras de DNA do tumor e de referência (reunidas a partir de sangue de diferentes doadores saudáveis; Promega, Madison, WI, EUA) foram diferentemente marcadas usando o kit de etiquetagem de DNA genômico (Enzo Genomic DNA Labeling), de acordo com as instruções do fabricante. Quinhentos ηg de DNA do tumor e de referência foram co-hibridados em um microarray de oligonucleotídeo 180K (SurePrint G3 Humano CGH Microarray Kit 4× 180K, modelo 22060. Agilent Technologies, Palo Alto, CA, EUA), de acordo com as intruções do fabricante. Esse modelo contém 24.011 sondas exônicas. Imagens dos microarrays foram obtidas pelo Agilent Microarray Scanner Bundle, e os dados foram extraídos usando o programa Feature Extraction v.9.1 (Agilent Technologies, Santa Clara, CA, EUA).
De dados do aCGH foram analisados com o programa Nexus Copy Number Discovery 7.0. As alterações genômicas no número de cópias foram identificadas com base no algoritmo de segmentação FASST2 (limiar de significância estabelecido em 5 × 10–8), com limiares da razão de log2 de 0,2 ou 0,8 para ganhos ou alto ganho de cópias, respectivamente, e –0,2 ou –1,0 para perdas ou perdas homozigóticas, respectivamente.
Resultados
Dados clínicopatológicos do CXAP derivado de APR
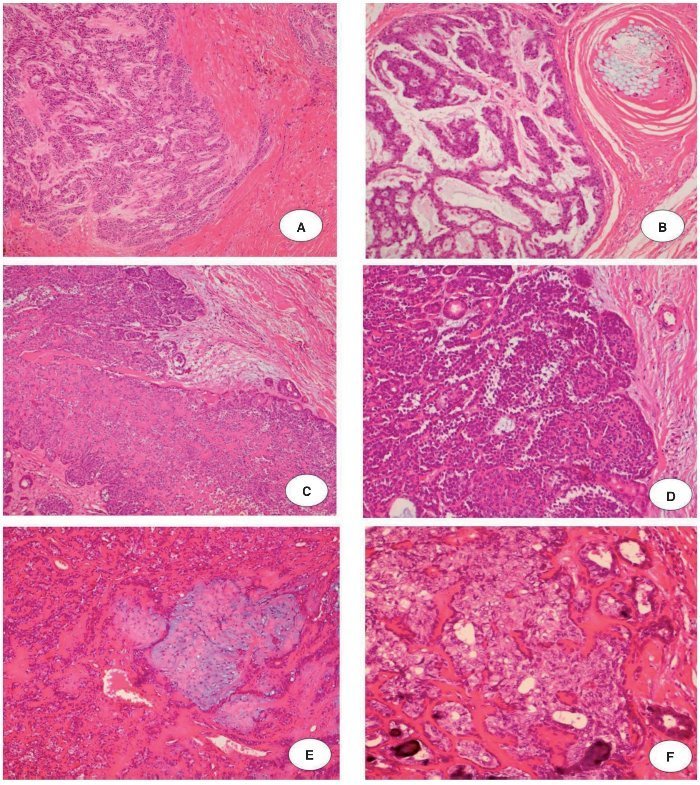
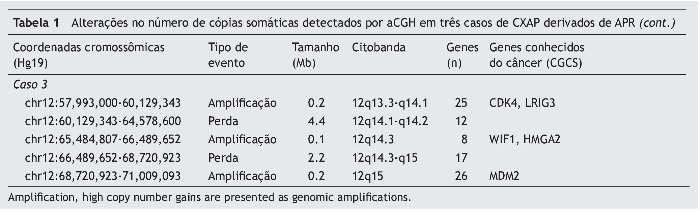
O primeiro paciente (caso 1) era um homem de 72 anos de idade, encaminhado ao nosso hospital para avaliação de um nódulo na glândula parótida medindo 9,0 × 8,0 centímetros, com um tempo relatado de evolução de dois anos. O paciente havia sido submetido à ressecção de um AP cinco anos antes. Durante o exame clínico, linfonodos palpáveis e invasão da pele subjacente foram observados. Havia ausência de lesões orais. Punção aspirativa por agulha fina revelou um AP. O tumor foi retirado com margens cirúrgicas positivas. O exame histológico mostrou a presença de AP e regiões de CXAP. A neoplasia foi classificada como um carcinoma mioepitelial francamente invasivo (fig. 1A e 1B). O paciente foi submetido à radioterapia e recorrência não foi observada em 58 meses de acompanhamento.
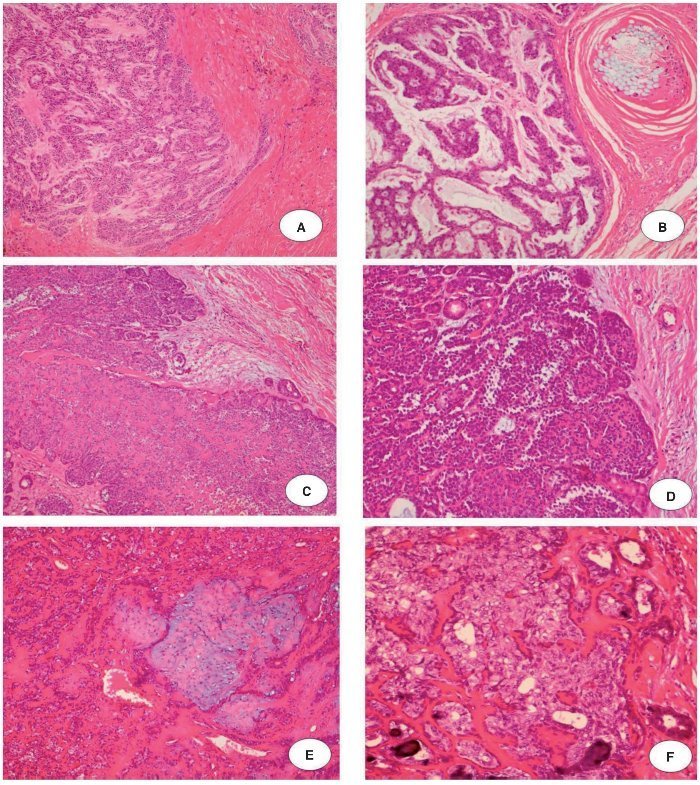
Figura 1. Carcinoma mioepitelial francamente invasivo: A, Ilha de células mioepiteliais infiltrando o tecido (H & E × 10); B, Cordões de células mioepiteliais pleomórficas cercado por estroma mixoide. Observe a reação contra a sutura de uma cirurgia anterior no lado superior direito da imagem (H & E × 20). Carcinoma epitelial-mioepitelial francamente invasivo: C, proliferação de células epiteliais e mioepiteliais em um crescimento nodular (H & E × 10); D, Pequeno lume delimitado por células eosinofílicas, cubóides, intercaladas, semelhantes aos do ducto. Essas células estão cercadas por pequenas células citoplasmáticas não coradas. Observe a manutenção de células basais na periferia de células ninho cercada por septos fibrosos (H & E × 20). Carcinoma epitelial-mioepitelial minimamente invasivo: E, Proliferação epitelial-mioepitelial provenientes de adenoma pleomórfico residual (H & E × 10); F, Material eosinofílico, hialinizado de lâmina basal circunda ninhos de células tumorais e estruturas ductais compostas de células epiteliais e mioepiteliais (H & E × 20).
A segunda paciente (caso 2) era uma mulher de 66 anos de idade, encaminhada ao nosso hospital com queixa de um tumor na região da parótida por tempo indeterminado. A paciente havia sido submetido à ressecção de um AP 11 anos antes. O exame clínico descartou a presença de linfonodos palpáveis e lesões orais. Punção aspirativa por agulha fina confirmou a presença de um AP. A excisão do tumor foi realizada, mas as margens eram cirurgicamente positivas. O exame histológico revelou a presença de AP e CXAP, que foram classificados como carcinoma epitelial-mioepitelial francamente invasivo (fig. 1C e 1D). Não houve acompanhamento dessa paciente.
A terceira paciente (caso 3) era uma mulher de 30 anos de idade, encaminhada ao nosso hospital com queixa de um tumor na glândula parótida com dois anos de duração. A paciente havia sido submetida à ressecção de um AP 16 anos antes. Durante o exame clínico, linfonodos palpáveis e lesões orais não foram observados. Punção aspirativa por agulha fina mostrou a presença de AP. O tumor foi excisado com margens cirúrgicas negativas. A análise histopatológica mostrou regiões de AP e CXAP. O último era um carcinoma epitelialmioepitelial minimamente invasivo (fig. 1E e 1F). Não houve acompanhamento dessa paciente.
Análise aCGH
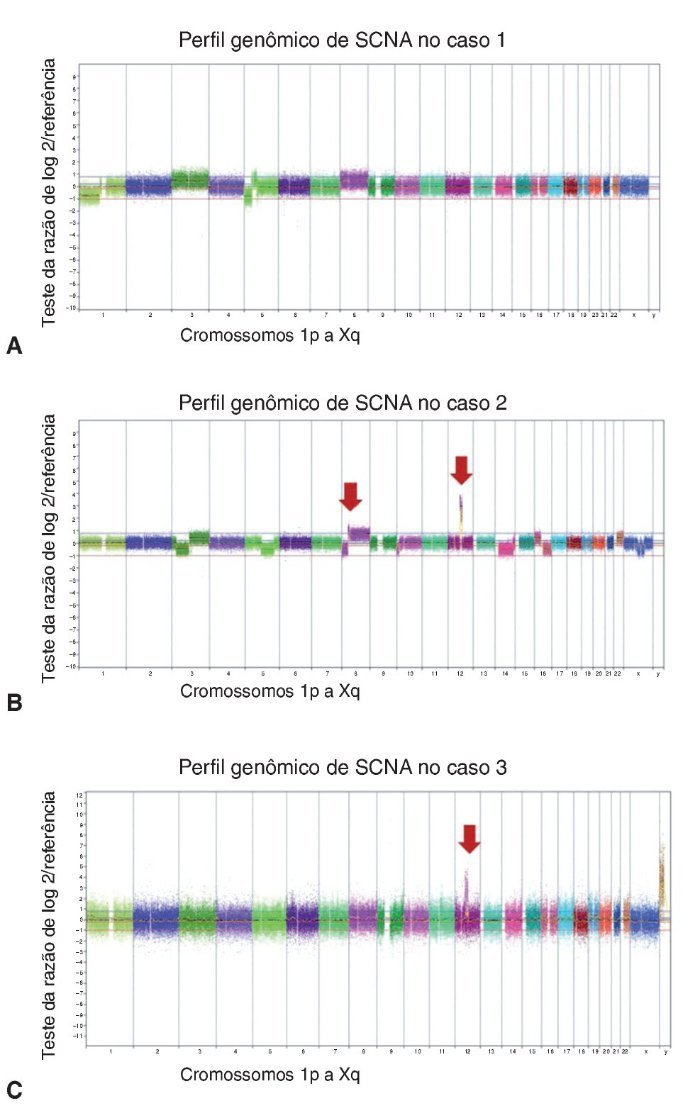
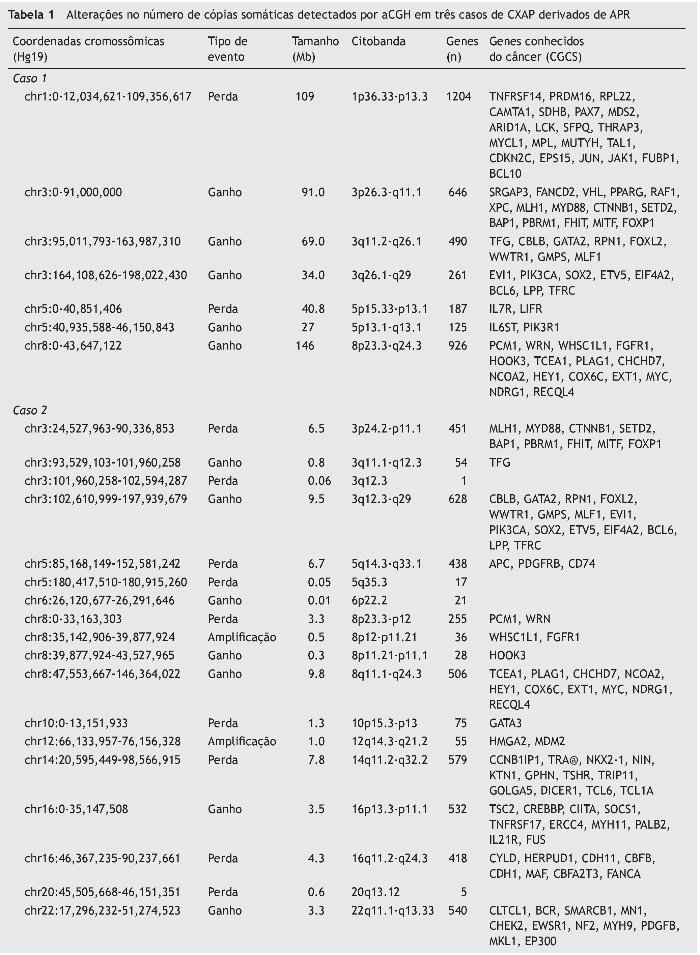
Os casos de APR não apresentaram alterações no número de cópias somáticas. Todas as alterações cromossômicas somáticas detectadas nos CXAP dos casos 1, 2 e 3 estão detalhadas na tabela 1, bem como os genes conhecidos do câncer afetados, de acordo com o Cancer Gene Census Sanger (https://www.sanger.ac.uk/research/projects/cancergenome/census.html). A figura 2 apresenta o perfil genômico global das alterações no número de cópias identificadas nos casos 1, 2 e 3 de CXAP. O primeiro CXAP derivado de APR exibiu perda em 1p36.33p13, ganhos nos cromossomos 8 e 3 e dois rearranjos cromossômicos adjacentes afetando 5p15.33p13.1 (perda) e 5p13.1q13.1 (ganho), respectivamente.
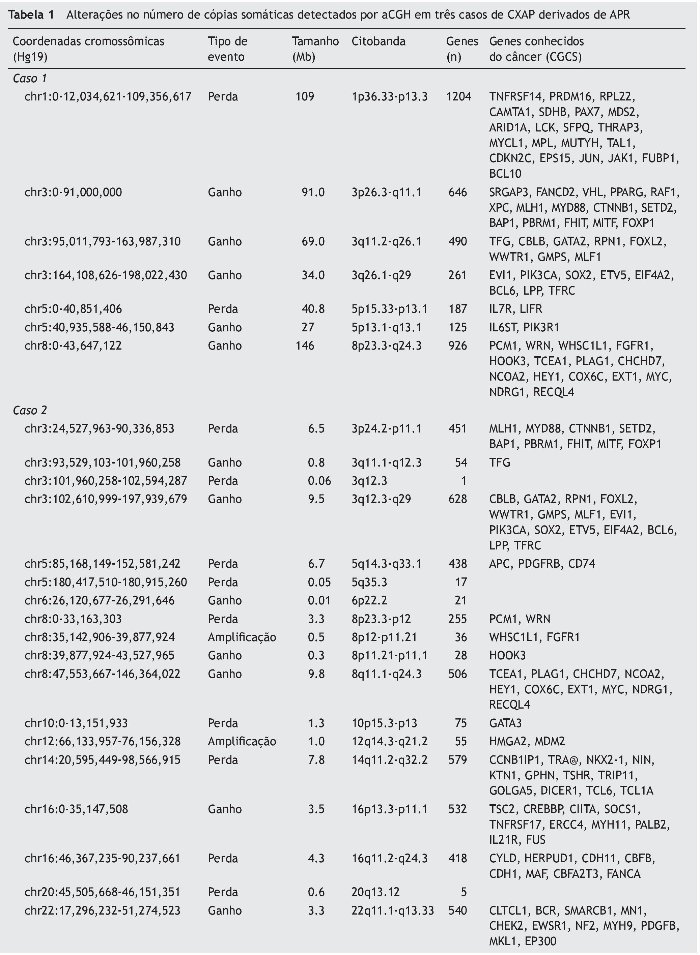
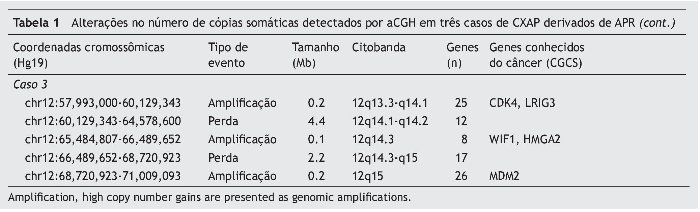
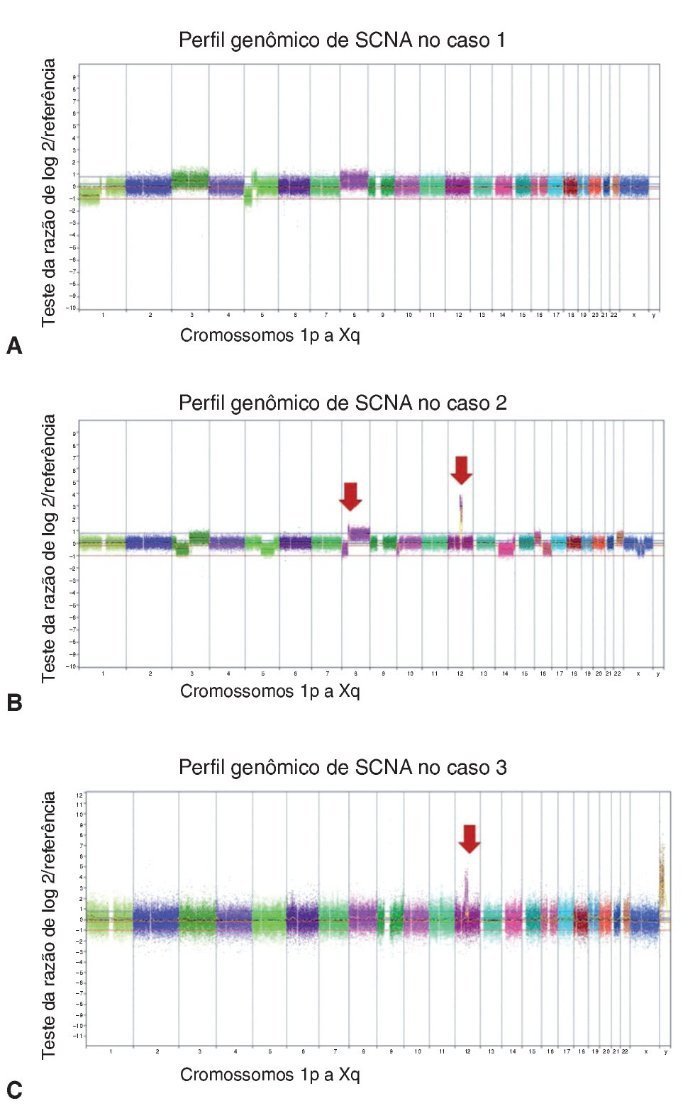
Figura 2. Alterações no número de cópias detectadas por aCGH em CXAPs derivados de APs. Perfil genômico por aCGH exibindo as alterações no número de cópias identificadas no caso de 1 (A), caso 2 (B) e caso 3 (C). O eixo x representa sondas ordenadas de acordo com suas posições genômicas a partir dos cromossomos 1p a Xq (cada cromossomo é marcado com uma cor diferente). O eixo y indica os valores de de referência/teste log2 (ganhos e perdas genômicas agrupados acima ou abaixo da linha de base 0, respectivamente; imagens adaptadas do programa Nexus Copy Number 7.0, Biodiscovery). As setas indicam o ganho elevado de cópias (amplificações).
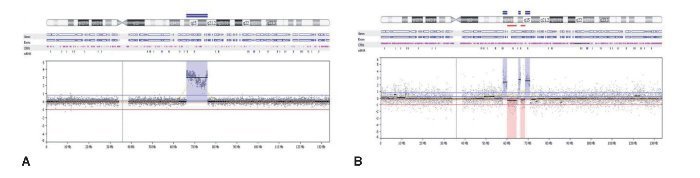
O segundo caso de CXAP derivado de APR apresentou um padrão genômico mais complexo, com várias alterações no número de cópias (ganhos e perdas), afetando os cromossomos 3, 8 e 16. Além disso, essa amostra incluiu perdas em 5q14.3q33.1, 5q35.3, 10p15. 3p13, 14q11.2q32.2, 20q13.12 e ganhos em 6p22.2, 22q11.1q13. Regiões de alto ganho no número de cópias (amplificações) foram encontrados em 8p12p11.21 e 12q14.3q21.2 (fig. 3A). Os genes amplificados incluíram, entre outros, WHSCILI e FGFR1 em 8p e HMGA2 e MDM2 em 12q.
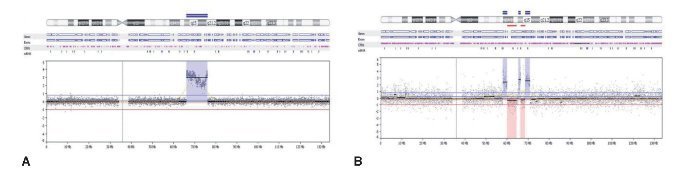
Figura 3. (A) Perfil aCGH do cromossomo 12 mostrando o ganho elevado no número de cópias (amplificação) de 1Mb em 12q14.3q21.2 no caso 1. (B) Perfil aCGH do cromossomo 12 mostrando um padrão complexo constituído por três regiões genômicas que apresentam elevado número de cópias (amplificações) em 12q13.3q14.1 (0,2 Mb), 12q14.3 (0,1 Mb) e 12q15 (0,2 Mb), interpolado com perdas no número de cópias de baixa amplitude.
O terceiro caso exibiu perdas em 12q14.1q14.2 e 12q14.3q15 e amplificações em 12q13.3q14.1, 12q.14.3 e 12q15, abrangendo CDK4, LRIG3, WIF1, HMGA2 e MDM2 (fig. 3B).
Discussão
A carcinogênese ocorre em várias etapas através de alterações genômicas que resultam em perda das funções supressoras do tumor, ativação de oncogenes e/ou geração de genes de fusão com potencial oncogênico.10 Essas alterações geram expansão clonal resultando no fenótipo de células cancerosas malignas.11
Embora tais alterações possam ocorrer por mutações ou rear-ranjos genômicos, números e estruturas anormais de cromossomos também foram documentados em células neoplásicas, indicando que a instabilidade cromossômica é um aspecto importante da biologia de células cancerosas.10 Portanto, as alterações no número de cópias podem ser uma ferramenta auxiliar na compreensão da carcinogênese.
A transformação maligna do AP recorrente foi relatada em 1,5% a 23%, e o risco parece aumentar com o tempo e o número de recidivas.12 A ocorrência de alteração maligna proveniente da recorrência do adenoma pleomórfico deve envolver a aquisição de mutações ao longo do tempo.
Os casos apresentados mostraram diferentes padrões de alterações no número de cópias, sendo importante ressaltar que as classificações histopatológicas e a característica invasiva também eram distintas. No entanto, a análise dos dados conseguiu identificar algumas alterações recorrentes no número de cópias, como ganhos em 3q e 8q (casos 1 e 2) e, principalmente, uma amplificação com uma região comum mínima em 12q14.3 (casos 2 e 3). Os casos 1 e 2 eram carcinomas francamente invasivos, e os ganhos em 3q e 8q podem estar implicados no grau de invasão. Os casos 2 e 3 eram carcinomas epiteliais-mioepiteliais, e a amplificação em 12q14.3 talvez esteja envolvida no subtipo histopatológico ou até na recorrência. O caso 2 exibiu um padrão mais complexo de rearranjos consistente com o subtipo histopatológico e o grau de invasão.
Perdas em 1p21.3-p21.1, 5q23.2-q31.2, 8p, 10q21.3 e 15q11.2 foram encontrados por Persson et al. (2009), em um estudo de um grupo de 10 CXAPs; no entanto, nenhuma classificação histopatológica foi realizada. Encontramos perdas em 1p36.33-p13, 5p15.33-P13.1, 5q14.3-q33.1, 5q35.3, 8p e 10p15.3-p13. Ganho em 8q12.1 (PLAG1), detectado em dois casos, foi relatado por vários autores.13-15
Amplificações de HMGA2, MDM2 e exclusões de 5q23.2q31.2 e 8q22.1q24.1 foram descritos como importante na transição do PA para CXAP.13 Observou-se alto ganho de cópias do HMGA2, MDM2, CDK4, WHSCIL1, LRIG3 e WIF1. Todos os genes amplificados são relacionados ao câncer, de acordo com o Cancer Gene Censo Sanger (https://www.sanger.ac.uk/research/projects/cancergenome/census.html). As amplificações de HMGA2 e MDM2 foram encontradas em dois de nossos casos, reforçando o seu papel como genes impulsionadores associados à recidiva no CXAP.
O gene HMGA2 (do grupo A humano de alta mobilidade) codifica uma proteína cromatina não histona que pertence a uma família de proteínas HMG, que são superexpressas em neoplasias malignas como do pulmão, pâncreas, carcinoma epidermóide de cavidade oral e câncer de mama.16-19 As proteínas HMGA2 possuem atividade oncogênica através de vários mecanismos como: indução de E2F1 e atividade de AP1, indução da expressão de ciclina A, inativação de p53 induzida por apoptose, impedimento da reparação do DNA, aumento da expressão de proteínas envolvidas na inflamação e modulação da expressão de microRNAs e genes envolvidos na transição epitelial-mesenquimal.20 Além disso, as proteínas HMGA têm um papel crucial na transformação celular porque, quando a sua síntese é bloqueada, ocorre a supressão do fenótipo de malignidade. Essa hipótese está de acordo com nosso achado, porque mostramos a amplificação de HMAG2 desde o caso minimamente invasivo.
A proteína MDM2 (murino duplo minuto-2 homólogo), também conhecida como ligase de proteína ubiquitina Mdm2 (E3), é um oncogene que codifica uma proteína Mdm2 que é um regulador negativo importante do supressor de tumor p53, degradando a proteína p53 ou inibindo a atividade de p53.21 A inibição de genes supressores de tumores ou a insensibilidade a sinais de anticrescimento ocorre na maioria dos tumores. As células cancerosas incipientes precisam evadir desses sinais antiproliferativos se quiserem prosperar.11 O presente trabalho mostrou a MDM2 já amplificada em nosso caso minimamente invasivo. A superexpressão de MDM2 também foi observada em uma ampla variedade de tumores humanos, como sarcoma, leucemia, carcinoma mamário, melanoma e glioblastoma.22
O WHSC1L1 (gene-1 candidato síndrome de Wolf-Hirschhorn) codifica uma pequena proteína contendo um domínio PWWP e é expresso em muitos tecidos. A função dessas proteínas codificadas não é clara, mas a presença do domínio PWWP, um local putativo para a interação proteína-proteína, sugere um papel regulador.23 O WHSC1L1 já foi identificada como um oncogene e é amplificado e superexpresso em carcinoma do pulmão.24
Os FGFR (receptores do fator de crescimento de fibroblastos), codificados por quatro genes (FGFR1, FGFR2, FGFR3, FGFR4) estão associados a muitos processos biológicos, como no desenvolvimento de órgãos e na proliferação e migração celular. Vários estudos relataram um papel dos FGFR na tumorigênese devido à regulação de diversos processos relacionados à tumorigênese, incluindo sobrevivência celular, proliferação, inflamação, angiogênese e metástase. A amplificação de FGFR1 foi identificada principalmente no câncer de pulmão.25
O CDK4 (cinases dependentes de ciclina 4) está diretamente envolvido na condução do ciclo celular (Lee et al., 2014). A amplificação de CDK4 foi observada em várias neoplasias malignas, incluindo glioma, câncer de mama, linfoma, melanoma e sarcoma. Às vezes, o CDK4 é coamplificado com MDM2. A proteína codificada por esse gene é uma subunidade catalítica do complexo proteína-cinase que é importante para a fase G1 de progressão do ciclo celular.26
A família de genes LRIG (domínios contendo repetições ricas em leucina e semelhantes à imunoglobulina) inclui: LRIG1, LRIG2 e LRIG3. A expressão de LRIG provou ser de valor prognóstico em diferentes tipos de cânceres humanos, incluindo câncer de mama, estágio inicial de câncer cervical espinocelular invasivo, carcinoma espinocelular cutâneo, oligodendroglioma e astrocitoma. LRIG1 funciona como um gene supressor tumoral, enquanto pouco se conhece sobre as funções de LRIG2 e LRIG3.27
WIF1 é um gene inibidor do fator de Wnt-1. A proteína codificada por este gene funciona para inibir as proteínas WNT, que são moléculas de sinalização extracelulares que desempenham um papel no desenvolvimento embrionário. Esse gene atua como gene supressor tumoral, e descobriu-se que é epigeneticamente silenciado em vários cânceres.28
Conclusão
Em conclusão, identificamos perfis genômicos exclusivos das alterações no número de cópias entre três casos de CXAP derivado de APR, e as diferenças podem ser explicadas devido aos subtipos histopatológicos e graus de invasão. No entanto, os ganhos recorrentes em 3q e 8q e amplificações em 12q14.3 e 12q15 detectados neste estudo podem ser os fatores promocionais de recidiva da doença.
Financiamento
Processo FAPESP: 2011/23204-5 e Processo FAPESP: 2011/23366-5.
Conflitos de interesse
Os autores declaram não haver conflitos de interesse.
Recebido em 11 de outubro de 2015;
aceito em 8 de dezembro de 2015
* Autor para correspondência.
E-mail:fevimariano@gmail.com (F.V. Mariano).
☆ Como citar este artigo: Mariano FV, Giovanetti K, Saccomani LF, Del Negro A, Kowalskic LP, Krepischi AC, et al. Carcinoma ex-pleomorphic adenoma derived from recurrent pleomorphic adenoma shows important difference by array CGH compared to recurrent pleomorphic adenoma without malignant transformation. Braz J Otorhinolaryngol. 2016;82:687-94.
DOI se refere ao artigo: http://dx.doi.org/10.1016/j.bjorl.2015.12.004


