



Several studies have associated congenital sensorineural hearing loss in children with prolongation of the cardiac parameter QTc. The cause of this association is unknown. At the same time, mutations in GJB2, which encodes connexin 26, are the most common cause of congenital hearing impairment.
ObjectiveTo compare electrocardiographic parameters (PR interval, QRS complex, and QTc interval) in patients with hearing loss who were tested for mutations in GJB2 and GJB6 to investigate whether these mutations affect electrical activity of the heart.
Methods346 patients (176 males, 170 females) with sensorineural hearing loss of 30dB HL or more, aged 21.8±19.9 years (including 147 children <14 years), underwent both genetic study for GJB2 and GJB6 mutations and electrocardiography.
ResultsMutations in GJB2, including homozygotes and heterozygotes, were found in 112 (32%) patients. There were no significant differences in ECG parameters between groups of patients with and without mutations in GJB2. No differences were observed either in men (mean PR with mutation: 155±16.6 vs. 153.6±30.1 without; QRS: 99.9±9.9 vs. 101.1±15.4; QTc: 414.9±29.9 vs. 412.4±25.7) or women (mean PR with: 148.7±21 vs. 143.8±22.8 without; QRS: 94.8±7.6 vs. 92.9±9.6; QTc: 416.8±20.6 vs. 424.9±22.8). In similar fashion, we did we find any significant differences between groups of children with and without GJB2 mutations (mean PR with: 126.3±19.6 vs. 127±19.7 without; QRS: 80.7±9.5 vs. 79.4±11.6; QTc: 419.7±23.5 vs. 419.8±24.8).
ConclusionNo association was found between the presence of GJB2 mutations encoding connexin 26 in patients with hearing loss and their ECG parameters (PR, QRS, QTc).
Vários estudos têm associado a perda auditiva neurossensorial congênita em crianças ao prolongamento do parâmetro cardíaco QTc. A causa dessa associação é desconhecida. Ao mesmo tempo, as mutações no GJB2, que codifica a conexina 26, são a causa mais comum de deficiência auditiva congênita.
ObjetivoComparar parâmetros eletrocardiográficos (intervalo PR, complexos QRS e intervalo QTc) em pacientes com perda auditiva que foram testados para mutações no GJB2 e GJB6 para investigar se essas mutações afetam a atividade elétrica do coração.
MétodoForam submetidos a estudo genético para mutações de GJB2 e GJB6 e eletrocardiograma 346 pacientes (176 homens, 170 mulheres) com perda auditiva neurossensorial de 30dB ou mais, com média de 21,8 ± 19,9 anos (incluindo 147 crianças <14 anos).
ResultadosMutações no GJB2, inclusive homozigóticos e heterozigóticos, foram encontradas em 112 (32%) pacientes. Não houve diferenças significativas nos parâmetros de ECG entre grupos de pacientes com e sem mutações no GJB2. Não foram observadas diferenças em homens (PR médio com mutação: 155 ± 16,6 vs. 153,6 ± 30,1 sem mutação; QRS: 99,9 ± 9,9 vs. 101,1 ± 15,4; QTc: 414,9 ± 29,9 vs. 412,4 ± 25,7) nem em mulheres (PR médio com: 148,7 ± 21 vs. 143,8 ± 22,8, sem; QRS: 94,8 ± 7,6 vs. 92,9 ± 9,6; QTc: 416,8 ± 20,6 vs. 424,9 ± 22,8). Da mesma forma, encontramos diferenças significativas entre os grupos de crianças com e sem mutações de GJB2 (PR médio com: 126,3 ± 19,6 vs. 127 ± 19,7, sem; QRS: 80,7 ± 9,5 vs. 79,4 ± 11,6; QTc: 419,7 ± 23,5 vs. 419,8 ± 24,8).
ConclusãoNão foi encontrada associação entre a presença de mutações de GJB2 que codificam conexina 26 em pacientes com perda auditiva e seus parâmetros de ECG (PR, QRS, QTc).
As conexinas são proteínas transmembranares integrais que formam canais na membrana celular, denominadas conexonas. Em células adjacentes, as conexonas formam junções estreitas que são necessárias para a transferência de sinais elétricos e moléculas hidrossolúveis. Elas ajudam a manter a homeostase do tecido e auxiliam no manejo e regulação do ambiente iônico de uma célula.1
As conexinas estão presentes em todo o corpo humano. Embora seja possível que células contenham números de diferentes conexinas, cada uma desempenha um papel distinto. Nos seres humanos, existem 21 proteínas que pertencem à família das conexinas e cada uma é codificada por um gene diferente.2 As mutações em genes que codificam as conexinas conduzem a distúrbios na sua estrutura e função, o que pode levar a doenças hereditárias, como surdez, catarata, doenças da pele e doenças neurodegenerativas.3–6
Nas células da orelha interna, a conexina 26 e a conexina 30 estão envolvidas em processos de transporte de íons de potássio necessários para a audição. Quando uma célula pilosa é estimulada, os íons de potássio na endolinfa entram nos cílios e no corpo da célula e fazem com que ela despolarize. Devido às conexões da junção de hiato, os íons são, por sua vez, transportados por células de suporte e células na estria vascular para a endolinfa. Por meio dessa recirculação de íons de potássio, um potencial endolinfático altamente positivo (+80mV) é gerado, uma condição necessária para a audição. A interrupção desse mecanismo provoca intoxicação do íon de potássio e dano tecidual.7
Tanto o gene GJB2 (MIM*121011) (que codifica a conexina 26) como o gene GJB6 (MIM*604418) (que codifica a conexina 30) estão localizados no cromossomo 13 (13q11‐q12). Mutações do gene GJB2 levam a distúrbios no funcionamento da conexina 26 e causam perda auditiva (PA) ao comprometer a transferência de íons de potássio intracelulares dentro da orelha interna.8 Mutações de GJB2 são uma das principais causas de PA não sindrômica autossômica recessiva em muitas populações. Entre os europeus, a mutação patogênica mais comum no gene GJB2 é uma deleção de guanina na posição 35 (NM_004004.5:c.35delG).9,10 Na maioria dos casos (70%), a PA ocorre como um defeito isolado; em outros, é acompanhada por defeitos cardíacos, craniofaciais, renais, cutâneos e outros.
Vários estudos relataram uma associação de PA neurossensorial congênita com prolongamento do intervalo QTc em crianças. A causa dessa associação é desconhecida.11,12
Este estudo mediu parâmetros eletrocardiográficos em pacientes com PA, que também foram testados para a mutação genética. A análise do eletrocardiograma (ECG) forneceu o intervalo PR, complexo QRS e intervalo QTc e esses parâmetros eletrocardiográficos foram comparados em dois grupos de pacientes: um com PA causada por mutações no gene GJB2 e outro com PA e nenhuma mutação nesse gene. Dessa maneira, foi possível investigar se havia uma relação entre as funções da conexina 26 e conexina 30 e atividade elétrica cardíaca.
MétodoA população de estudoO grupo de estudo consistiu em 346 pacientes consecutivos, com média de 21,8±19,9 anos (0,16‐84 anos), inclusive 176 homens e 170 mulheres, com PA neurossensorial de 30dB ou mais (calculada como o limiar auditivo médio a frequências relevantes para a fala ‐ 0,5; 1; 2 e 4kHz), nos quais foram feitos testes genéticos para identificar as mutações em GJB2 e GJB6. A PA foi confirmada por audiometria tonal pura ou, no caso de crianças com menos de 6 anos, com potencial evocado auditivo do tronco cerebral (BERA). Entre os indivíduos estudados, 91 tinham implantes cocleares.
Testes genéticosO DNA foi extraído do sangue periférico por meio de um procedimento de salting‐out.13 A tipagem para a mutação c.35delG no gene GJB2 foi feita por dois métodos independentes: AS‐PCR e PCR‐RFLP, tal como descrito anteriormente.14,15 Em resumo, foi feita a amplificação multiplex de três regiões de GJB2 (c.12‐72, c.68‐198 e c.306‐464) com primers marcados fluorescentes. Em paralelo, foi feita análise de PCR‐RFLP específica para mutação c.35delG. O tamanho dos fragmentos gerados pelos dois protocolos anteriormente mencionados foi determinado por meio de um analisador genético 3500XL (Life Technologies), com um software mapeador genético (Life Technologies). As amostras que não tinham duas mutações c.35delG foram ainda analisadas por sequenciação direta do DNA do exon 2 amplificado por PCR do gene. A análise da sequência foi feita com o software Variant Reporter v1.1 (Life Technologies). A genotipagem da mutação c‐23+1G>A de GJB2 foi feita com PCR em tempo real, de acordo com as instruções do fabricante (Life Technologies), enquanto a mutação de GJB6 del (GJB6‐D13S1830) foi detectada com o método de del Castillo et al.16 A nomenclatura genética está alinhada com as recomendações da Human Genome Variation Society (HGVS).
Exames de eletrocardiogramaTodos os 346 pacientes foram submetidos a eletrocardiograma (ECG) de 12 derivações. O registro do ECG foi feito e avaliado com o sistema Sentinel Cardiology Information Management (Reynolds Medical). As durações de intervalo PR, complexo QRS e intervalo QTc foram medidas por um único observador, em uma imagem aumentada 5 vezes em um monitor de computador com uma precisão de cursor de 1ms (frequência de amostragem 1000Hz). O intervalo PR foi geralmente medido em derivação II, complexo QRS em V1 e/ou V5, e intervalo QT em pelo menos três derivações do mesmo ciclo (geralmente derivações II, V2 e V5). A frequência cardíaca foi calculada a partir do intervalo RR que precede o ciclo do intervalo QT medido. Para corrigir o intervalo QT devido à frequência cardíaca (isto é, QTc), a fórmula de Bazett foi usada para frequências entre 50 e 120 bpm; para frequências mais rápidas ou mais lentas, foi usada a fórmula de Hodges. Para calcular QTc médio, foram excluídos os pacientes com complexos QRS ≥ 120ms. Um intervalo QTc acima de 460ms nas mulheres, 450ms nos homens e 440ms em crianças com menos de 14 anos foi diagnosticado como prolongado.
A população do estudo foi dividida em três grupos, com base em valores de referência do QTc para crianças, mulheres e homens,17 ficaram 147 crianças<14 anos (42,4% dos pacientes), 100 mulheres ≥ 14 anos (28,9%) e 99 homens ≥ 14 anos (28,6%). O grupo de crianças incluiu 70 (47%) meninas.
Análise estatísticaA análise estatística de cada um dos três grupos envolveu um teste de Kruskal‐Wallis e o teste de duas amostras de Wilcoxon.
OutrosO estudo foi aprovado pelo Comitê de Bioética, número KB/05/2009. O estudo foi feito em conformidade com os princípios da Declaração de Helsinque. Todos os pacientes ou seus responsáveis forneceram consentimento escrito para participar.
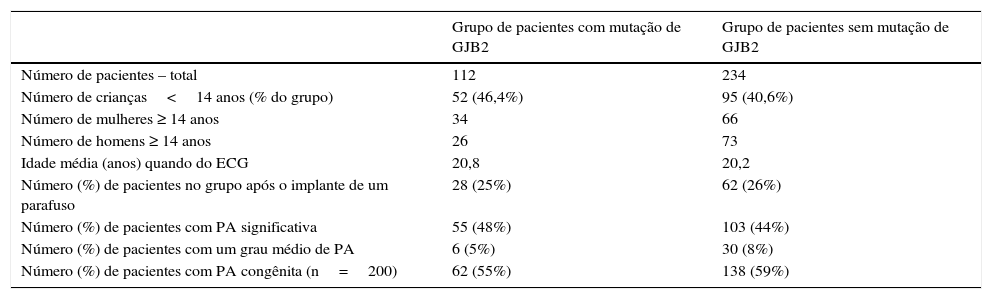
ResultadosCaracterísticas da população de estudo e estudos genéticosAs mutações em GJB2 foram identificadas em 112 (32%) pacientes com PA. Nenhuma mutação foi detectada nos genes GJB2 e GJB6 analisados nos outros 234 (68%) pacientes.
A idade média de crianças com uma mutação GJB2 e crianças sem mutação GJB2 foi semelhante (6,0±4,6 vs. 5,8±4,5 anos, p=0,86). A idade média dos adultos nos dois grupos (com e sem mutação de GJB2) também foi semelhante (35,1±17,6 vs. 30,5±16,5 anos, p=0,17 para as mulheres; 31,6±18,9 e 30,3±17,5 anos, p=0,99 para os homens) (tabela 1).
Características do grupo de pacientes diagnosticados com mutações no gene GJB2 (genótipo homozigótico e heterozigótico juntos) e o grupo de pacientes sem qualquer mutação no gene
| Grupo de pacientes com mutação de GJB2 | Grupo de pacientes sem mutação de GJB2 | |
|---|---|---|
| Número de pacientes – total | 112 | 234 |
| Número de crianças<14 anos (% do grupo) | 52 (46,4%) | 95 (40,6%) |
| Número de mulheres ≥ 14 anos | 34 | 66 |
| Número de homens ≥ 14 anos | 26 | 73 |
| Idade média (anos) quando do ECG | 20,8 | 20,2 |
| Número (%) de pacientes no grupo após o implante de um parafuso | 28 (25%) | 62 (26%) |
| Número (%) de pacientes com PA significativa | 55 (48%) | 103 (44%) |
| Número (%) de pacientes com um grau médio de PA | 6 (5%) | 30 (8%) |
| Número (%) de pacientes com PA congênita (n=200) | 62 (55%) | 138 (59%) |
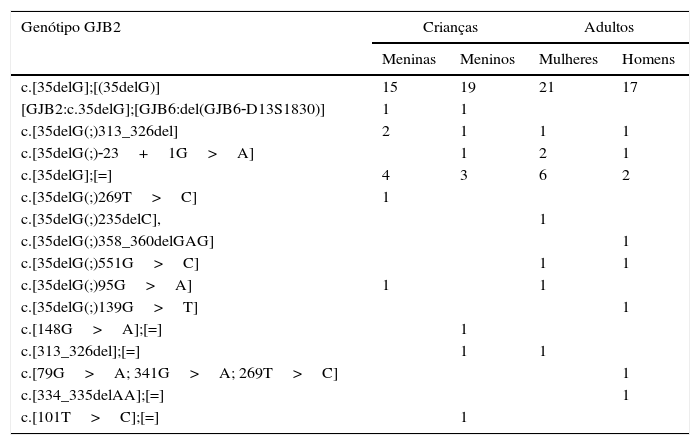
O genótipo homozigoto c.[35delG];[(35delG)] no gene GJB2 foi detectado em 72 (20,8%) indivíduos. Em 40 (11,8%) indivíduos, foram identificados genótipos GJB2 heterozigotos: c.[35delG]; [=] (n=15), c.[35delG (;) 313_326del] (n=5), c.[35delG (;) ‐23+1G>A] (n=4), c.[313_326del];. [=] (n=2), c.[35delG (;) 551G>C] (n=2), c.[35delG (;) 95G>a] (n=2), e um genótipo heterozigoto composto de GJB2 e GJB6 [GJB2:c.35delG]; [GJB6:del(GJB6‐D13S1830)] (n=2). Os seguintes genótipos heterozigotos de GJB2 ocorreram em casos individuais: c.[35delG(;) 269T>C], c.[148g>A]; [=], c.[79g>A; 341G>A; 269T>C], c.[101T>C];. [=], c.[35delG.(;)235delC], c.[35delG.(;)358_360delGAG], c.[35delG.(;)139g>T], c.[334_335delAA]; [=] (tabela 2).
Tipos de genótipo e sua distribuição na população do estudo
| Genótipo GJB2 | Crianças | Adultos | ||
|---|---|---|---|---|
| Meninas | Meninos | Mulheres | Homens | |
| c.[35delG];[(35delG)] | 15 | 19 | 21 | 17 |
| [GJB2:c.35delG];[GJB6:del(GJB6‐D13S1830)] | 1 | 1 | ||
| c.[35delG(;)313_326del] | 2 | 1 | 1 | 1 |
| c.[35delG(;)‐23+1G>A] | 1 | 2 | 1 | |
| c.[35delG];[=] | 4 | 3 | 6 | 2 |
| c.[35delG(;)269T>C] | 1 | |||
| c.[35delG(;)235delC], | 1 | |||
| c.[35delG(;)358_360delGAG] | 1 | |||
| c.[35delG(;)551G>C] | 1 | 1 | ||
| c.[35delG(;)95G>A] | 1 | 1 | ||
| c.[35delG(;)139G>T] | 1 | |||
| c.[148G>A];[=] | 1 | |||
| c.[313_326del];[=] | 1 | 1 | ||
| c.[79G>A; 341G>A; 269T>C] | 1 | |||
| c.[334_335delAA];[=] | 1 | |||
| c.[101T>C];[=] | 1 | |||
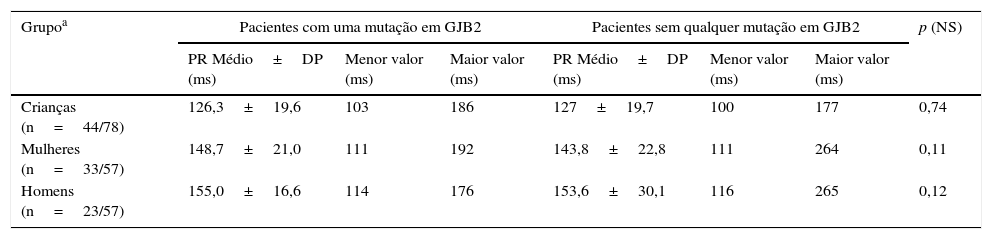
A próxima fase do estudo foi comparar parâmetros eletrocardiográficos na população estudada. Os parâmetros medidos foram comparados entre os dois grupos: o grupo de pacientes com uma mutação no gene GJB2 (ambos homozigotos e heterozigotos) e o grupo de pacientes sem mutação no gene. Não foram encontradas diferenças significativas para qualquer parâmetro medido em qualquer um dos grupos estudados. A tabela 3 apresenta as diferenças intergrupos no intervalo PR; a tabela 4 apresenta as diferenças intergrupos no complexo QRS; e a tabela 5 apresenta diferenças intergrupo no intervalo QTc.
Achados de intervalo PR no ECG da população do estudo
| Grupoa | Pacientes com uma mutação em GJB2 | Pacientes sem qualquer mutação em GJB2 | p (NS) | ||||
|---|---|---|---|---|---|---|---|
| PR Médio±DP (ms) | Menor valor (ms) | Maior valor (ms) | PR Médio±DP (ms) | Menor valor (ms) | Maior valor (ms) | ||
| Crianças (n=44/78) | 126,3±19,6 | 103 | 186 | 127±19,7 | 100 | 177 | 0,74 |
| Mulheres (n=33/57) | 148,7±21,0 | 111 | 192 | 143,8±22,8 | 111 | 264 | 0,11 |
| Homens (n=23/57) | 155,0±16,6 | 114 | 176 | 153,6±30,1 | 116 | 265 | 0,12 |
Achados de complexo QRS no ECG da população do estudo
| Grupo | Pacientes com uma mutação em GJB2 | Pacientes sem uma mutação em GJB2 | p (NS) | ||||
|---|---|---|---|---|---|---|---|
| QRS médio±DP (ms) | Menor valor (ms) | Maior valor (ms) | QRS médio±DP (ms) | Menor valor (ms) | Maior valor (ms) | ||
| Crianças (n=49/89)a | 80,7±9,5 | 65 | 99 | 79,4±11,6 | 61 | 114 | 0,32 |
| Mulheres (n=33/59) | 94,8±7,6 | 80 | 110 | 92,9±9,6 | 65 | 118 | 0,31 |
| Homens (n=23/59) | 99,9±9,9 | 85 | 119 | 101,1±15,4 | 65 | 156 | 0,98 |
Achados de intervalo QTc no ECG da população do estudo
| Grupoa | Pacientes com uma mutação em GJB2 | Pacientes sem uma mutação em GJB2 | p (NS) | ||||
|---|---|---|---|---|---|---|---|
| QTc médio±DP (ms) | Menor valor (ms) | Maior valor (ms) | QTc médio±DP (ms) | Menor valor (ms) | Maior valor (ms) | ||
| Crianças (52/94) | 419,7±23,5 | 366 | 479 | 419,8±24,8 | 371 | 483 | 0,91 |
| Mulheres (34/64) | 416,8±20,6 | 371 | 456 | 424,9±22,8 | 370 | 467 | 0,08 |
| Homens (26/61) | 414,9±29,9 | 366 | 485 | 412,4±25,7 | 362 | 476 | 0,86 |
Além disso, foram comparados os parâmetros em pacientes com PA profunda causada por uma mutação de GJB2 de genótipo homozigoto c.[35delG];[(35delG)] e naqueles com um genótipo heterozigoto [GJB2: c.35delG];[GJB6: del(GJB6‐D13S1830)] vs. todos os outros pacientes com outros tipos de PA. A análise não revelou diferenças significativas (tabela 6).
Achados de intervalo PR, complexo QRS e intervalo QTc em dois grupos. Grupo A, pacientes com genótipo homozigótico c.[35delG];[(35delG)] e genótipo heterozigótico [GJB2:c.35delG];[GJB6:del(GJB6‐D13S1830)]; Grupo B, Todos os outros pacientes (com outro genótipo heterozigótico e sem qualquer mutação em GJB2)
| Crianças (n=31/90)a | Mulheres (n=20/70) | Homens (n=15/58) | |
|---|---|---|---|
| PR médio±DP (ms) no grupo A | 122,55±18,51 | 144,1±22,98 | 153,8±15,07 |
| PR médio±DP (ms) no grupo B | 128,02±25,38 | 146,3±25,74 | 154,05±25,55 |
| p | 0,0633 | 0,3934 | 0,1174 |
| Crianças (n=33/104) | Mulheres (n=20/72) | Homens (n=15/60) | |
|---|---|---|---|
| QRS médio±DP (ms) no grupo A | 79,81±9,81 | 94,05±7,91 | 98,6±10,18 |
| QRS médio±DP (ms) no grupo B | 79,87±13,2 | 93,24±13,17 | 101,27±13,88 |
| p | 0,4528 | 0,4124 | 0,4577 |
| Crianças (n=36/111) | Mulheres (n=20/79) | Homens (n=17/74) | |
|---|---|---|---|
| QTc médio±DP (ms) no grupo A | 418,33±20,4 | 417,3±18,45 | 412,41±32,65 |
| QTc médio±DP (ms) no grupo B | 420,5±24,16 | 423,74±24,07 | 414,47±24,16 |
| p | 0,4462 | 0,0908 | 0,4594 |
Finalmente, analisou‐se a incidência de anomalias no ECG no grupo de pacientes com uma mutação no GJB2 e naqueles sem qualquer mutação deste gene. As anormalidades no grupo de pacientes com uma mutação foram: ritmo atrial (dois pacientes), bradicardia sinusal (n=12), condução atrioventricular de tempo curto (n=3), bloqueio fascicular anterior esquerdo (n=1), intervalo QTc acima da norma para a idade e sexo (n=10) e sinais de necrose da parede inferolateral do miocárdio associados ao prolongamento do QTc (n=1). No grupo de pacientes sem qualquer mutação, as anormalidades foram: bradicardia sinusal (n=25), ritmo atrial (n=3), marcapasso atrial migratório (n=1), tempo curto de condução atrioventricular (n=2), bloqueio atrioventricular de primeiro grau (n=5), bloqueio de ramo incompleto direito (n=1), bloqueio fascicular anterior esquerdo (n=1), bloqueio de ramo completo (n=7), distúrbios da condução intraventricular (n=3), sinais de hipertrofia ventricular esquerda (n=3), sinais de isquemia do miocárdio (n=2) e prolongamento do QTc (n=23). Em um menino, havia ritmo supraventricular com um tempo de condução atrioventricular prolongado, distúrbios de condução intraventricular inespecíficos e sinais de hipertrofia ventricular direita.
No grupo de pacientes com mutação de GJB2 e QTc prolongado (n=10), o intervalo QTc médio foi de 463,2±15,6ms, enquanto nos pacientes com prolongamento do QTc e sem qualquer mutação (n=23), o intervalo QTc médio foi de 460,3±12,8ms. Essa diferença também não foi significativa (p=0,69).
DiscussãoNosso estudo obteve ECG de 346 pacientes com PA que foram testados para mutações nos genes GJB2 e GJB6. Há uma ausência geral de dados sobre parâmetros de ECG detalhados em populações grandes semelhantes. Há relatos de ECG normais feitos em 11 pacientes entre 9 e 20 anos, em cinco famílias com PA associada a mutações de GJB2.18 Além disso, outro estudo19 descreveu 270 crianças com PA neurossensorial, dos quais 143 foram submetidos a um ECG: foram detectadas anomalias em dez (7%), três tiveram intervalo QT prolongado (uma teve múltiplos defeitos atriais e septais ventriculares e duas foram diagnosticadas com síndrome de Jervell e Lange‐Nielsen) e outras anomalias relatadas incluíram bradicardia sinusal, bloqueio de ramo direito e hipertrofia ventricular esquerda. De 220 crianças no grupo de estudo, 33 (15%) tinham mutações em ambos os alelos do gene GJB2.
Este estudo é o primeiro de que temos conhecimento e que descreve os parâmetros de ECG, como intervalo PR e complexo QRS, em um grande grupo de pacientes com PA. As mutações no gene GJB2 causam PA em 20‐60% das crianças caucasianas. A prevalência de mutações de GJB2 na população europeia é mais elevada nos países do Mediterrâneo e mais baixa no norte da Europa. Estima‐se que a mutação c.35delG é portada por 1% a 3% da população europeia.20,21 O genótipo de GJB2 mais comum que ocorre em pacientes europeus com PA é o homozigoto c.[35delG];[(35delG)].20,21 No nosso grupo de estudo de pacientes com PA, esse genótipo foi também o mais comum. Além disso, mostramos que entre a população estudada há também outras mutações, embora elas sejam raras. Uma mutação rara, a c.167delT relatada por Dong et al.,22 deve ser observada; nota‐se que essa mutação é rara na Europa em geral, mas muito mais frequente em judeus Ashkenazi.23 Em contraste, a mutação mais comum em japoneses é c.235delC, em que a sua prevalência é de 1% ‐2,1%.24
Além da conexina 26, um papel biológico semelhante na orelha interna também recebe contribuição da conexina 30, codificada pelo gene GJB6, cujo locus está localizado na proximidade imediata do gene GJB2 no cromossomo 13.25,26 A deleção de 342kb que abrange o gene GJB6 foi descrita, particularmente em pacientes espanhóis. Outros estudos mostraram que os efeitos fenotípicos de mutações em ambos os genes são idênticos e um genótipo heterozigótico composto [GJB2:c.35delG];[GJB6: del(GJB6‐D13S1830)] é caracterizado por perda de audição.16
A nossa análise dos parâmetros eletrocardiográficos em pacientes com PA causada por mutação de GJB2 mostrou que a condução atrioventricular (definida pelo intervalo PR), o tempo de condução intraventricular (definido pela duração de QRS) e o intervalo QTc não diferiram significativamente em comparação com os pacientes com PA que não tinham mutação nesse gene.
Limitações do estudoNosso estudo teve algumas limitações. Diferentes expressões fenotípicas são resultado de várias mutações de GJB2. Como as mutações do gene GJB2 descritas que resultam em PA são recessivas, comparamos os parâmetros de um grupo de pacientes com um genótipo homozigótico c.[35delG];[(35delG)] e um heterozigótico [GJB2: c.35delG];[GJB6:del(GJB6‐D13S1830)] com todos os outros pacientes. Os resultados dessa análise são limitados pelo pequeno número de pessoas com um genótipo homozigótico c.[35delG];[(35delG)] e havia apenas duas crianças com genótipos heterozigotos [GJB2: c.35delG];[GJB6:del (GJB6‐D13S1830)] dando o efeito fenotípico da surdez.
No grupo de estudo, a presença de defeitos cardíacos não foi descartada por um teste confiável (p.ex., ecocardiografia) e nem tampouco o efeito de determinados medicamentos que podem ter influenciado parâmetros eletrocardiográficos. O grupo de estudo foi selecionado pela triagem de indivíduos com PA que não relataram sintomas de doença cardíaca ou não tomavam medicamentos, o que produziu um grupo aparentemente saudável que apresentava PA.
ConclusãoEm pacientes com PA, não foi encontrada associação entre os parâmetros de ECG e a presença de mutações de GJB2 que codificam conexina 26.
Conflitos de interesseOs autores declaram não haver conflitos de interesse.
Este trabalho foi apoiado pelo National Center for Science [número de concessão NN 402526739].
A revisão por pares é da responsabilidade da Associação Brasileira de Otorrinolaringologia e Cirurgia Cérvico‐Facial.
Como citar este artigo: Sanecka A, Biernacka EK, Sosna M, Mueller‐Malesinska M, Ploski R, Skarzynski H, et al. Evaluation of electrocardiographic parameters in patients with hearing loss genotyped for the connexin 26 gene (GJB2) mutations. Braz J Otorhinolaryngol. 2017;83:176–82.
gology tem o prazer em homenagear os revisores


