
Nasopharyngeal carcinoma is the most common cancer originating from the nasopharynx.
ObjectiveTo study the mechanisms of nasopharyngeal carcinoma, we analyzed GSE12452 microarray data.
MethodsGSE12452 was downloaded from the Gene Expression Omnibus database and included 31 nasopharyngeal carcinoma samples and 10 normal nasopharyngeal tissue samples. The differentially expressed genes were screened by ANOVA in the PGS package. Using the BiNGO plugin in Cytoscape and pathway enrichment analysis in the PGS package, functional and pathway enrichment analyses were performed separately to predict potential functions of the differentially expressed genes. Furthermore, Transcription factor‐differentially expressed gene pairs were searched, and then the transcription factor‐differentially expressed gene regulatory network was visualized using Cytoscape software.
ResultsA total of 487 genes were screened as differentially expressed genes between the nasopharyngeal carcinoma samples and the normal nasopharyngeal tissue samples. Enrichment analysis indicated that PTGS2 was involved in the regulation of biological process and small cell lung cancer. ZIC2 and OVOL1 may function in nasopharyngeal carcinoma through targeting significantly up‐regulated genes (such as PTGS2, FN1, CXCL9 and CXCL10) in the Transcription factor‐differentially expressed gene regulatory network (e.g., ZIC2→PTGS2 and OVOL1→CXCL10).
ConclusionPTGS2, FN1, CXCL9, CXCL10, ZIC2 and OVOL1 might play roles in nasopharyngeal carcinoma.
O carcinoma nasofaríngeo é o câncer mais comum originário da nasofaringe.
ObjetivoEstudar os mecanismos do câncer de nasofaringe; dados do microarray GSE12452 foram analisados.
MétodoGSE12452 foi obtido da base de dados Gene Expression Omnibus e inclui 31 amostras de carcinoma nasofaríngeo e 10 amostras de tecido nasofaríngeo normal. Os genes diferencialmente expressos foram analisados por ANOVA no kit PGS. Usando o plugin BiNGO no Cytoscape e análise de enriquecimento da via no kit PGS, análises de enriquecimento funcional e da via foram realizadas separadamente para prever as potenciais funções dos genes diferencialmente expressos. Além disso, os pares Fator de Transcrição - genes diferencialmente expressos foram pesquisados e em seguida a sua rede reguladora foi visualizada usando o programa Cytoscape.
ResultadosUm total de 487 genes foram analisados como genes diferencialmente expressos entre as amostras de carcinoma nasofaríngeo e amostras de tecido nasofaríngeo normal. A análise de enriquecimento indicou que PTGS2 estava envolvido na regulação do processo biológico e câncer pulmonar de pequenas células. ZIC2 e OVOL1 podem funcionar no carcinoma nasofaríngeo almejando‐se de maneira significativa os genes suprarregulados (como o PTGS2, FN1, CXCL9 e CXCL10) na rede reguladora de fator de transcrição - genes diferencialmente expressos (p.ex., ZIC2→PTGS2 e OVOL1→CXCL10).
ConclusãoPTGS2, FN1, CXCL9, CXCL10, ZIC2 e OVOL1 podem desempenhar alguns papéis no carcinoma de nasofaringe.
O câncer de nasofaringe mais comum, o carcinoma nasofaríngeo (CNF), causou 86.700 novos casos e 50.800 mortes no mundo em 2012.1 O CNF é extremamente comum no sudeste da Ásia e no sul da China, com mais de 50.000 novos casos por ano,2,3 e pode ser induzido por vários fatores, como hereditariedade, fatores virais e influências ambientais.4 A maioria dos casos de CNF está correlacionada à infecção pelo vírus Epstein‐Barr (EBV), um herpesvírus B‐linfotrópico que tem propriedades de transformação de crescimento.5 Assim, o estudo dos mecanismos do CNF é de grande importância.
Muitos estudos que investigam os mecanismos de CNF foram publicados. Existem vários genes (como C‐myc, AKT1, p53, MDM2, LMP1 e PTEN) implicados na patogenia do CNF, porque eles são muitas vezes amplificados ou alterados em pacientes com essa doença.6–8 O Disabled 2 (DAB2) é frequentemente infrarregulado por hipermetilação do promotor e pode ser um supressor tumoral potencial no CNF.9 Estudos anteriores mostram que o potencial gene supressor tumoral de uma desintegrina e metaloprotease com motivos trombospondina 9 tipo 1 (ADAMTS9) está estreitamente relacionado com metástases para linfonodos e pode inibir o crescimento do tumor por meio da supressão da angiogênese no CNF.10,11 O complexo 1da proteína relacionada com o adaptador (AP‐1) do fator de transcrição (TF) ativado pelo antígeno nuclear 1 codificado pelo EBV (EBNA 1) pode ter como alvo o fator‐1α induzido por hipóxia, interleucina 8 e fator de crescimento endotelial vascular (VEGF), que promove a formação de microtúbulos em células do CNF.12 O TF forkhead Box M1 (FOXM1) está envolvido no desenvolvimento do tumor e o vetor do adenovírus AdFOXM1shRNA, que expressa short hairpin RNA específico de FOXM1, pode ser usado como intervenção terapêutica para o tratamento de pacientes com CNF.13 Por infrarregulação da expressão da proteína secretada, ácida e rica em cisteína (SPARC), a região de determinação do sexo do TF Y‐box 5 (SOX‐5) atua na progressão do CNF e pode ser usado como um preditor para o prognóstico sombrio de CNF.14,15 Embora esses estudos tenham sido feitos para investigar CNF, seus mecanismos ainda permanecem obscuros.
Em 2006, Sengupta et al. analisaram a expressão de todos os genes de EBV latentes entre amostras de CNF e amostras de epitélio da nasofaringe normais e obtiveram um painel de genes diferencialmente expressos (GDE).3 Com os mesmos dados empregados por Sengupta et al.,3 não só foram selecionados os GDE, mas também foi feita uma análise bioinformática abrangente para identificar os principais genes associados ao CNF. As funções potenciais dos GDE foram previstas por análises funcionais e da via de enriquecimento. Além disso, uma rede reguladora TF‐GDE foi construída para investigar as relações de regulação entre TF e GDE.
MétodoDados de microarrayOs dados do perfil de expressão de GSE12452 depositados por Sengupta et al.3 foram obtidos do banco de dados Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/), que se baseou na plataforma do GPL570 [HG‐U133_Plus_2] Affymetrix Human Genome U133 Plus 2.0 Array. Os espécimes incluíram 31 amostras de CNF e 10 amostras de tecido de nasofaringe normais coletadas de pacientes de Taiwan, com consentimento informado. As amostras foram ressecadas, rapidamente congeladas e armazenadas em nitrogênio líquido.
Triagem para GDEPara o conjunto de dados GSE12452, o método Robust MultiArray Averaging (RMA) no kit Partek® Genomics Suite™ (PGS) (http://www.partek.com/)16 foi usado para pré‐processamento, inclusive o ajuste de fundo, a normalização de quantil e a transformação log2. Após a correção do lote, foi obtida a matriz de expressão processada. O método Anova (análise de variância) no kit PGS16 foi usado para comparar os GDE entre as amostras de CNF e as de tecidos normais da nasofaringe. Os valores de p obtidos foram ajustados para testes múltiplos com o método de Benjamini & Hochberg.17 Genes com fold‐change (FC)>2 e valor p ajustado<0,05 foram selecionados como GDE.
Análise funcional e de via de enriquecimentoOs termos de ontologia do gene (OG) podem ser usados para descrever os três tipos de produtos de genes, inclusive o processo biológico no qual eles estão implicados, sua função molecular e localização subcelular.18 A Kyoto Encyclopedia of Genes and Genomes (KEGG) é um banco de dados composto por genes e suas informações funcionais.19 Com o plug‐in BiNGO20, em Cytoscape (http://www.cytoscape.org/), e análise de enriquecimento da via no kit PGS,16 análises de enriquecimento da via GO e KEGG foram feitas separadamente para GDE entre as amostras de CNF e amostras de tecidos normais da nasofaringe. O valor p ajustado<0,05 foi usado como o critério de corte.
Construção da rede reguladora TF‐GDEO pacote de software Genomatix Suite (https://www.genomatix.de/)21 foi usado para prever fatores de transcrição (TF). Em resumo, a ferramenta Gene2Promoter (http://www.genomatix.de)22 foi usada para extrair sequências promotoras de GDE correspondentes a partir da base de dados Genomatix. Em seguida, os TF foram analisados com a ferramenta MatInspector.21 TF com um valor de p<0,05 e FC>2 foram selecionados como TF expressos diferencialmente, e, em seguida, os seus correspondentes pares de TF‐GDE foram analisados. Finalmente, a rede reguladora TF‐GDE foi visualizada com o software Cytoscape.23
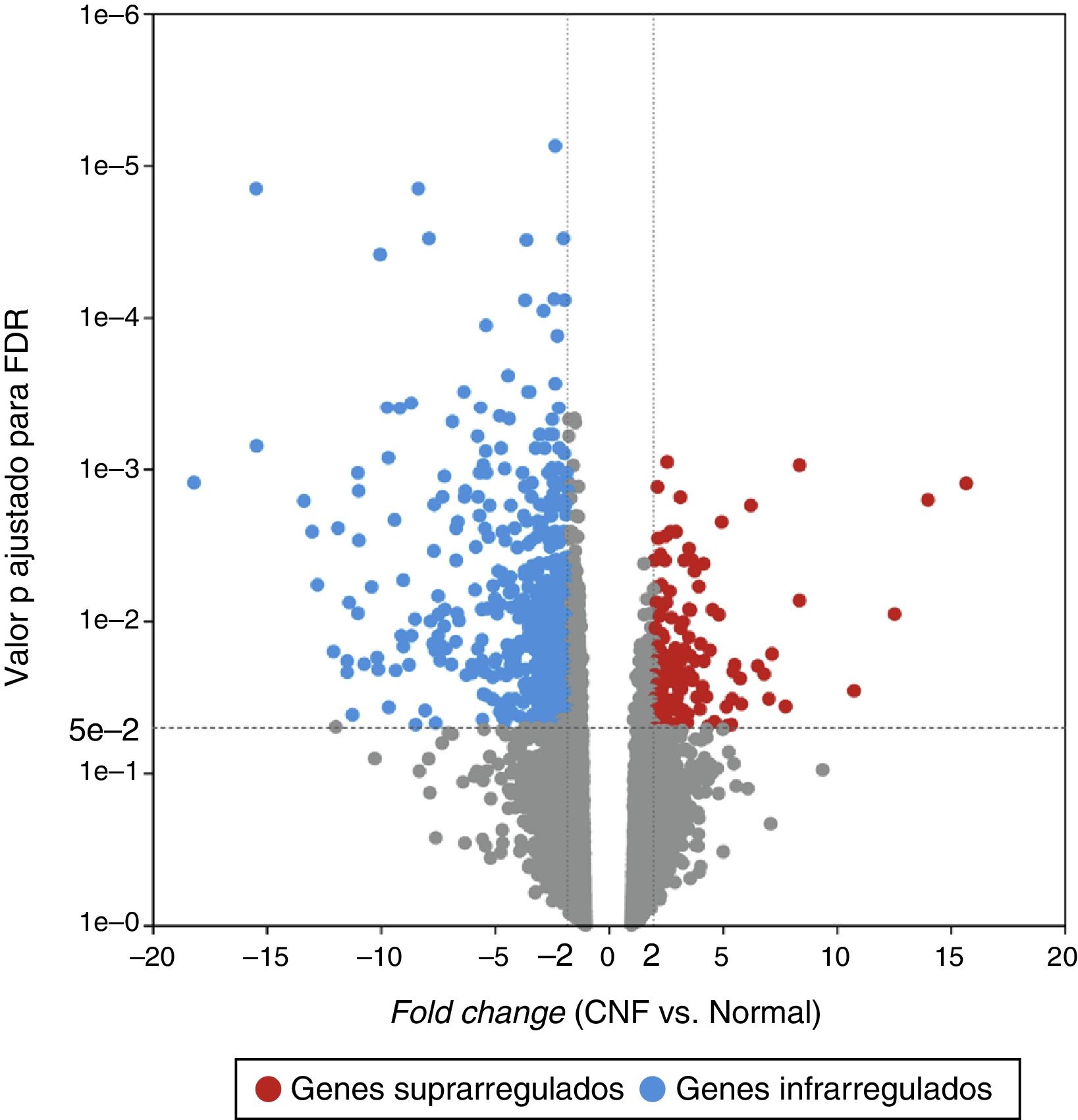
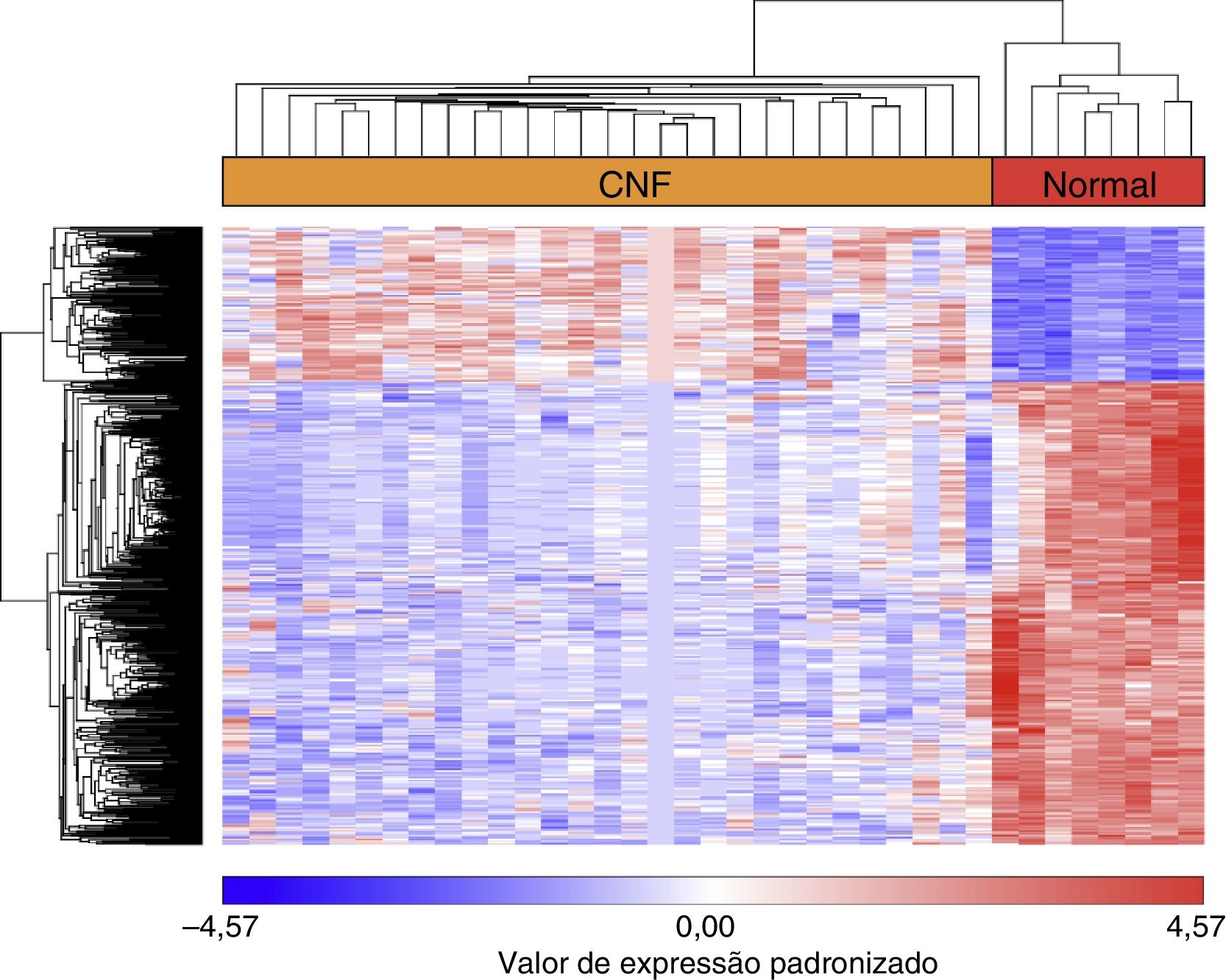
ResultadosAnálise de GDEApós o GSE12452 ser baixado, os dados de perfil de expressão foram pré‐processados e, em seguida, os GDE foram identificados por Anova com o kit PGS. O gráfico volcano para GDE, separadamente, é mostrado na figura 1. Foram selecionados 487 genes como GDE entre as amostras de CNF e as amostras de tecidos nasofaríngeos normais, inclusive 122 genes suprarregulados e 365 infrarregulados. O número de genes infrarregulados foi maior do que o número de genes suprarregulados. No heat map da análise hierárquica de agrupamento para os GDE, as amostras do CNF e as amostras de tecidos normais da nasofaringe eram claramente divididas em dois grupos (fig. 2).
 (FC>2 e valor p ajustado<0,05). O eixo horizontal representa o fold change e o eixo vertical representa o valor p ajustado. Os círculos vermelhos e azuis indicam genes suprarregulados e infrarregulados, respectivamente.")
.")
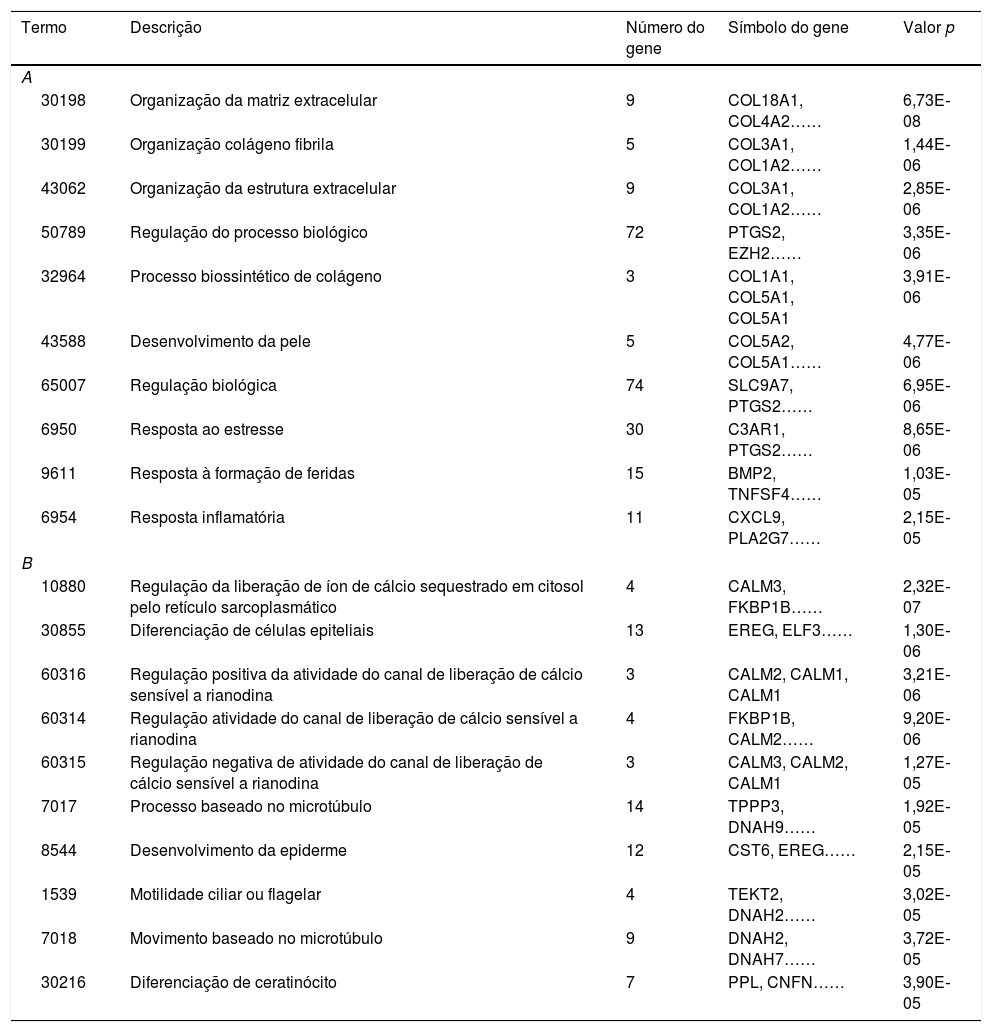
As dez principais funções de GO para os genes suprarregulados incluíram a organização de fibrilas de colágeno (p=1,44E‐06), a organização de matriz extracelular (p=6,73E‐08) e regulação do processo biológico (p=3,35E‐06, que envolveu prostaglandina‐endoperóxido sintase 2, PTGS2) (tabela 1A). As vias KEGG enriquecidas para genes suprarregulados estão listadas na tabela 1C e incluíram amebíase (p=1,25E‐07), interação ECM‐receptor (p=9,01E‐10) e câncer de pulmão de pequenas células (p=3,10E‐06, que envolveu PTGS2).
Da mesma maneira, análises de enriquecimento funcionais e de via foram feitas para os genes infrarregulados. Como pode ser visto na tabela 1B, as funções que incluem a regulação da liberação do íon de cálcio sequestrado para o citosol por retículo sarcoplasmático (p=2,32E‐07), diferenciação de células epiteliais (p=1,30E‐06) e regulação positiva de atividade do canal de liberação de cálcio sensível a rianodina (p=3,21E‐06) foram enriquecidas. As vias enriquecidas incluíram citocromo P450 do metabolismo do fármaco (p=3,02E‐07), metabolismo de xenobióticos por citocromo P450 (p=8,48E‐06) e metabolismo da tirosina (p=1,69E‐04) (tabela 1D).
Função enriquecida de GO e as vias de KEGG para os genes suprarregulados e infrarregulados. (A) As dez principais funções do GO enriquecidas para os genes suprarregulados. (B) As dez principais funções do GO enriquecidas para os genes infrarregulados. (C) As dez principais vias de KEGG enriquecidas para os genes suprarregulados. (D) As dez principais vias de KEGG enriquecidas para os genes infrarregulados
| Termo | Descrição | Número do gene | Símbolo do gene | Valor p |
|---|---|---|---|---|
| A | ||||
| 30198 | Organização da matriz extracelular | 9 | COL18A1, COL4A2…… | 6,73E‐08 |
| 30199 | Organização colágeno fibrila | 5 | COL3A1, COL1A2…… | 1,44E‐06 |
| 43062 | Organização da estrutura extracelular | 9 | COL3A1, COL1A2…… | 2,85E‐06 |
| 50789 | Regulação do processo biológico | 72 | PTGS2, EZH2…… | 3,35E‐06 |
| 32964 | Processo biossintético de colágeno | 3 | COL1A1, COL5A1, COL5A1 | 3,91E‐06 |
| 43588 | Desenvolvimento da pele | 5 | COL5A2, COL5A1…… | 4,77E‐06 |
| 65007 | Regulação biológica | 74 | SLC9A7, PTGS2…… | 6,95E‐06 |
| 6950 | Resposta ao estresse | 30 | C3AR1, PTGS2…… | 8,65E‐06 |
| 9611 | Resposta à formação de feridas | 15 | BMP2, TNFSF4…… | 1,03E‐05 |
| 6954 | Resposta inflamatória | 11 | CXCL9, PLA2G7…… | 2,15E‐05 |
| B | ||||
| 10880 | Regulação da liberação de íon de cálcio sequestrado em citosol pelo retículo sarcoplasmático | 4 | CALM3, FKBP1B…… | 2,32E‐07 |
| 30855 | Diferenciação de células epiteliais | 13 | EREG, ELF3…… | 1,30E‐06 |
| 60316 | Regulação positiva da atividade do canal de liberação de cálcio sensível a rianodina | 3 | CALM2, CALM1, CALM1 | 3,21E‐06 |
| 60314 | Regulação atividade do canal de liberação de cálcio sensível a rianodina | 4 | FKBP1B, CALM2…… | 9,20E‐06 |
| 60315 | Regulação negativa de atividade do canal de liberação de cálcio sensível a rianodina | 3 | CALM3, CALM2, CALM1 | 1,27E‐05 |
| 7017 | Processo baseado no microtúbulo | 14 | TPPP3, DNAH9…… | 1,92E‐05 |
| 8544 | Desenvolvimento da epiderme | 12 | CST6, EREG…… | 2,15E‐05 |
| 1539 | Motilidade ciliar ou flagelar | 4 | TEKT2, DNAH2…… | 3,02E‐05 |
| 7018 | Movimento baseado no microtúbulo | 9 | DNAH2, DNAH7…… | 3,72E‐05 |
| 30216 | Diferenciação de ceratinócito | 7 | PPL, CNFN…… | 3,90E‐05 |
| C | |||
| Interação ECM‐receptor | 11 | COL1A1, COL1A2…… | 9,01E‐10 |
| Digestão e absorção da proteína | 10 | COL1A2, COL3A1…… | 9,05E‐09 |
| Amebíase | 10 | CD14, COL1A2…… | 1,25E‐07 |
| Câncer de pulmão de pequenas células | 8 | PTGS2, FN1…… | 3,10E‐06 |
| Infecção por Staphylococcus aureus | 5 | C3AR1, FCGR2A…… | 2,66E‐04 |
| Diferenciação de osteoclastos | 6 | SOCS1, TYK2…… | 2,23E‐03 |
| Via de sinalização do receptor Toll‐like | 5 | CXCL9, NFKBIA…… | 4,54E‐03 |
| Reprodução do DNA | 3 | PCNA, MCM2…… | 6,36E‐03 |
| Leishmaniose | 4 | FCGR2A, PTGS2…… | 6,94E‐03 |
| Ciclo celular | 5 | CCNE2, SMC1A…… | 9,53E‐03 |
| D | |||
| Metabolismo do fármaco‐citocromo P450 | 9 | ADH1C, ADH7…… | 3,02E‐07 |
| Metabolismo de xenobióticos por citocromo P450 | 8 | ADH7, ALDH1A3…… | 8,48E‐06 |
| Metabolismo da tirosina | 5 | ALDH3A1, ALDH3B2…… | 1,69E‐04 |
| Glicólise/gliconeogênese | 6 | ADH7, ALDH1A3…… | 2,51E‐04 |
| Metabolismo da histidina | 4 | ALDH3A1, ALDH3A2…… | 5,74E‐04 |
| Metabolismo de beta‐alanina | 4 | ALDH3A2, ALDH3B2…… | 6,59E‐04 |
| Metabolismo de fenilalanina | 3 | ALDH1A3, ALDH3A1, ALDH3B2 | 1,88E‐03 |
| Junção de oclusão | 7 | CGN, CLDN7…… | 2,40E‐03 |
| Fototransdução | 3 | CALM1, CALM2, CALM3 | 7,52E‐03 |
| Metabolismo do retinol | 4 | DHRS9, SDR16C5…… | 1,03E‐02 |
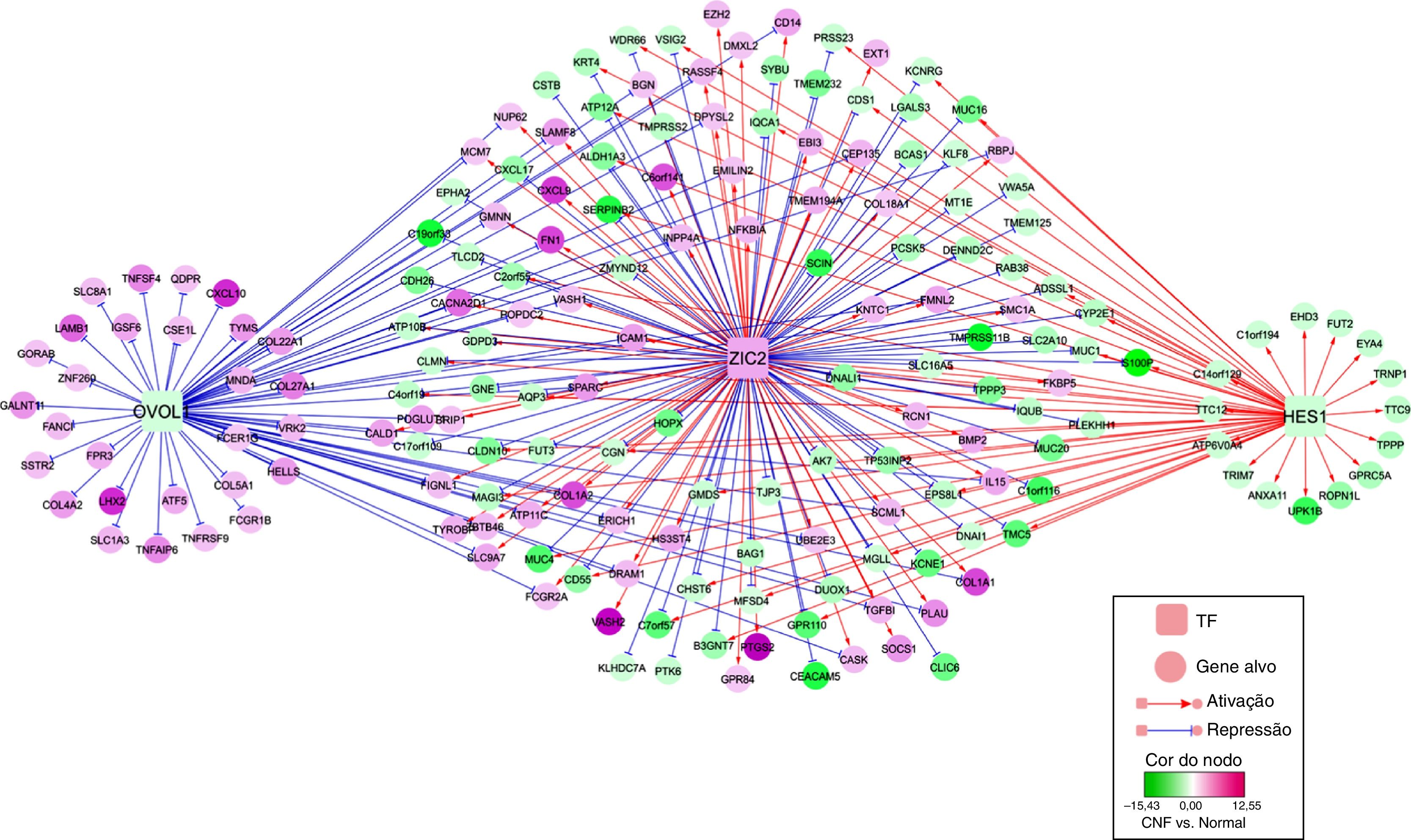
Com o pacote Genomatix Software Suite, os TF foram previstos para 122 genes suprarregulados e 365 genes infrarregulados. Havia TF suprarregulados que almejavam genes suprarregulados ou infrarregulados, bem como TF infrarregulados que almejavam gentes suprarregulados e infrarregulados. No entanto, apenas TF que tiveram como alvo tanto genes suprarregulados como infrarregulados foram selecionados. A rede reguladora de TF‐GDE dos TF (membroda família Zic 2, ZIC2; tipo ovo 1, OVOL1; e o hairy/enhancer of split 1, HES1) e seus GDE alvo são mostrados na figura 3. Em particular, PTGS2 significativamente suprarregulado, fibronectina (FN1) e quimiocinas (CXC motif) que ligam 9 (CXCL9) foram almejados e ativados por ZIC2. No entanto, quimiocina significativamente suprarregulada (C‐X‐C motif) que liga 10 (CXCL10) foi almejada e suprimida por OVOL1.
Discussão e seus genes diferencialmente expressos almejados (GDE).")
Neste estudo, foram selecionados 487 genes como amostras GDE entre amostras de CNF e amostras de tecidos nasofaríngeos normais, inclusive 122 genes suprarregulados e 365 infrarregulados. Havia muitas funções enriquecidas para os GDE, como a organização de matriz extracelular e organização da estrutura extracelular. PTGS2, FN1, CXCL9 e CXCL10 eram genes significativamente suprarregulados, envolvidos na rede reguladora TF‐DEG, e podem funcionar no CNF através de ZIC2 ou OVOL1 (p.ex., ZIC2 → PTGS2 e OVOL1 → CXCL10).
A análise de enriquecimento indicou que PTGS2 estava envolvido na regulação do processo biológico e câncer de pulmão de pequenas células. Como um gene a jusante envolvido na via de NF‐κB, PTGS2 também é chamado de ciclo‐oxigenase‐2 (COX‐2). Sabe‐se que COX2 pode regular células cancerosas do tipo estaminais das células de CNF e promover as suas características. Além disso, o partenolídeo pode funcionar na quimioterapia dirigida por meio da via do NF‐κB/COX2.24 Com base em PCR e análise de polimorfismo de comprimento de fragmentos de restrição, o polimorfismo −765 G> do promotor funcional C do COX2 pode ser relacionado a risco e progressão neoplásica de CNF.25 Os resultados de imuno‐histoquímica e avaliação semiquantitativa mostram que a expressão de COX‐2 aumenta à medida que o epitélio da nasofaringe evolui de normal a displásico e, em seguida, para CNF, indica que COX‐2 promove o desenvolvimento de CNF.26,27 Isso sugere que PTGS2 pode estar associado ao CNF.
Sabe‐se que FN1 contribui para a tumorigênese por meio da promoção do crescimento e migração de células tumorais, bem como do aumento da resistência à terapia.28,29 O FN1 suprarregulado correlaciona‐se com a transição epitelial para mesenquimatosa (TEM) e contribui para a metástase de células tumorais; pode, portanto, ser usado como um potencial marcador de diagnóstico e alvo terapêutico de CNF.30 Com base no sistema de arranjo de suspensão multiplex, um estudo anterior descobriu que a expressão de CXCL9 é significativamente suprarregulada em pacientes com CNF e carcinoma espinocelular da cavidade oral.31,32 A PCR quantitativa em tempo real e a imuno‐histoquímica sugeriram que CXCL9 suprarregulado está relacionado com agressividade do CNF e um ensaio imunoabsorvente ligado a enzima indicou que seu nível sérico pode ser um indicador de prognóstico valioso.33 Os genes ZIC, que consistem em cinco domínios de dedo de zinco Cys2His2, codificam fatores de transcrição de dedos de zinco.34 Entre os membros da família, ZIC2 medeia a expressão específica de tecido do receptor da dopamina D1 acoplado à proteína G.35 A expressão de ZIC2 é suprarregulada em diversos tumores malignos, inclusive sarcoma sinovial, meduloblastoma pediátrica e cânceres do endométrio.36–38 Portanto, os níveis de FN1, CXCL9 e ZIC2 podem estar correlacionados com o CNF. Na rede reguladora TF‐DEG, descobrimos que ZIC2 almejou e ativou FN1, CXCL9 e PTGS2, indica que ZIC2 pode também funcionar em CNF e regular FN1, CXCL9 e PTGS2.
Relata‐se que os TF OVOL1 e OVOL2 desempenham papéis cruciais na indução da transição de mesenquimatoso para epitelial (TME) em vários tipos de cânceres humanos.39 Como um membro da família quimioquina CXC, CXCL10 liga‐se ao seu receptor CXCR3 para exercer funções biológicas em doenças infecciosas, disfunção imune, inflamação crônica, desenvolvimento de tumores e metástases.40 A quimiocina CXC negativa para ELR CXCL10 pode atenuar a angiogênese e suprimir tumores.41 A sobre‐expressão de CXCL10 e CXCR3 desempenha papéis essenciais em cânceres avançados, tais como carcinoma de células basais,42 linfoma de células B,43 mieloma múltiplo44 e carcinoma do ovário.45 As citocinas inflamatórias que incluem CXCL10, proteína inibidora de macrófago 1 (MIP1) e interleucina 1 alfa são sobre‐expressadas em células malignas de CNF, que podem promover o infiltrado de leucócitos.46 Esses resultados sugerem que OVOL1 e CXCL10 podem estar correlacionados com CNF. Na rede reguladora TF‐DEG, também descobrimos que OVOL1 tinha como alvo e reprimia CXCL10, sugere que OVOL1 pode também desempenhar um papel na mediação do CNF por CXCL10.
ConclusãoForam usados os dados do perfil de expressão de GSE12452 baixados da GEO para estudar os mecanismos de CNF. Foram selecionados 487 genes como GDE entre as amostras de CNF e as de tecidos normais da nasofaringe. Vários genes (PTGS2, FN1, CXCL9, CXCL10, ZIC2 e OVOL1) podem desempenhar papéis no CNF e também podem ser usados para a terapia alvo de CNF na prática clínica. Entretanto, uma validação experimental adicional ainda é necessária para desvendar seus mecanismos de ação no CNF.
Conflitos de interesseOs autores declaram não haver conflitos de interesse.
Como citar este artigo: Jiang X, Feng L, Dai B, Li L, Lu W. Identification of key genes involved in nasopharyngeal carcinoma. Braz J Otorhinolaryngol. 2017;83:670–6.
A revisão por pares é da responsabilidade da Associação Brasileira de Otorrinolaringologia e Cirurgia Cérvico‐Facial.
gology tem o prazer em homenagear os revisores


