
O neurofibroma plexiforme solitário é uma forma muito rara de tumor, principalmente na porção parotídea do nervo facial. Os diagnósticos diferenciais são principalmente neurofibromas e outros tumores da parótida e a diversidade de suas apresentações clínicas e radiológicas são o principal motivo de desafios diagnósticos e terapêuticos.1–3
Relato de casoUm homem de 56 anos apresentou uma lesão tumoral parotídea esquerda de aumento progressivo por 2 anos. Nenhuma dor foi observada, nenhuma assimetria facial ou fraqueza. Não havia história de trauma anterior ou de neurofibromatose endócrina múltipla tipo 1 (NF1), no histórico médico pessoal ou familiar. Além disso, nenhuma intervenção anterior relevante foi relatada.
A lesão tinha 2cm de largura e 2cm de largura, era aderente a estruturas profundas e superficiais, firme, adjacente ao ângulo mandibular inferiormente e ao lóbulo da orelha superiormente. A ressonância magnética (RM) da região da parótida mostrou um tumor semelhante ao adenoma cístico linfático da glândula parótida. O paciente estava ciente do risco de comprometimento do nervo facial e a cirurgia foi planejada.
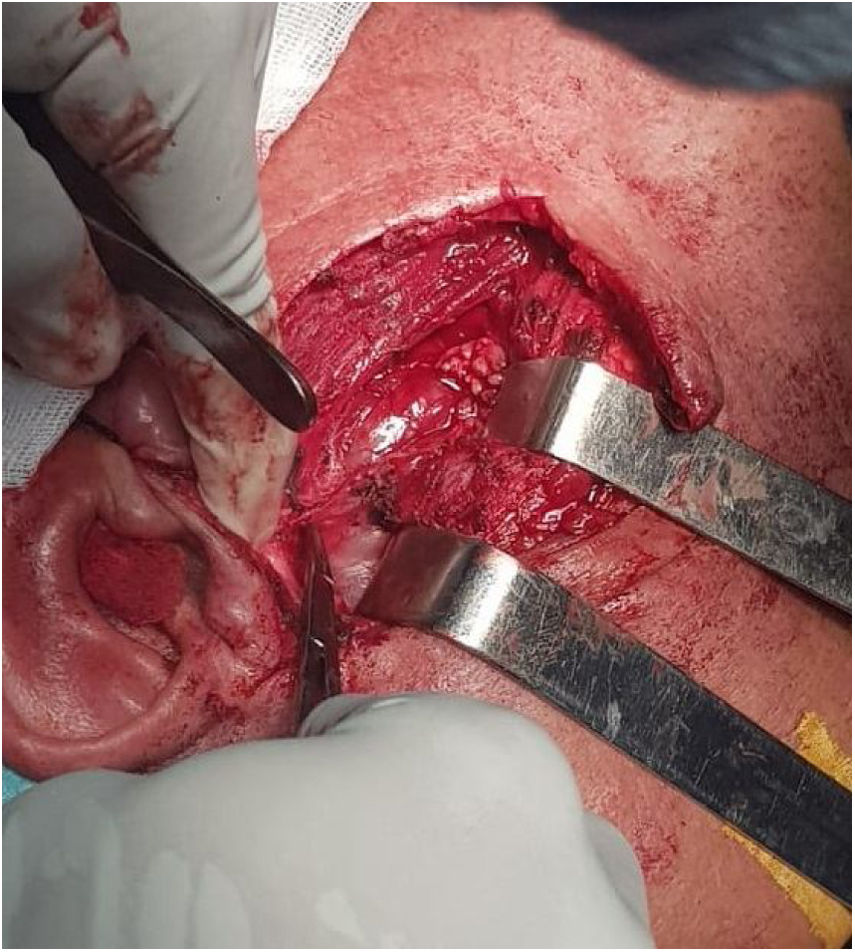
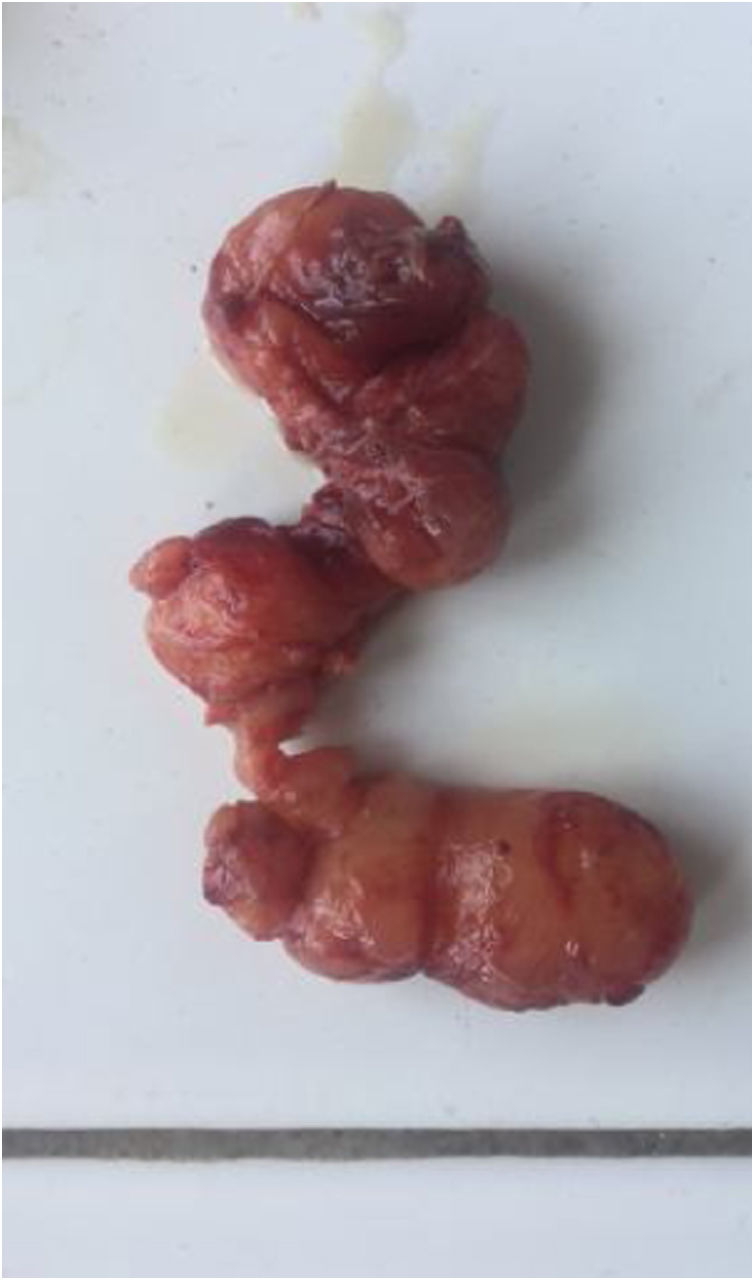
A exploração perioperatória encontrou tecido saudável nos tecidos cutâneo, subcutâneo e parotídeo; a lesão originava‐se do ponto de emergência do tronco do nervo facial, na base do processo estiloide (fig. 1) e estendia‐se até a porção intraparotídea, até a borda anterior do músculo masseter. A excisão completa do tumor implicou a excisão do nervo facial, com poucas fibras íntegras (fig. 2). A reparação cirúrgica do nervo não foi possível. O resultado pós‐operatório imediato mostrou grau quatro de paralisia do nervo facial periférico na escala de House‐Brackmann.


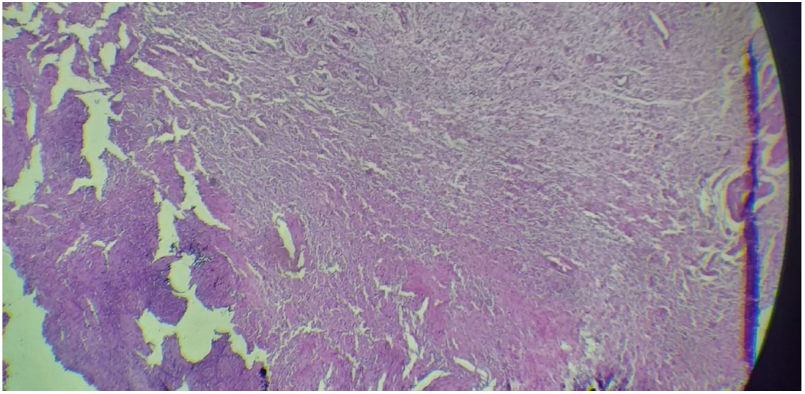
O aspecto histológico mostrou que o tumor não era encapsulado: ele incluía uma mistura de células de Schwann, células perineurais e fibroblastos endoeurais (fig. 3). O diagnóstico de neurofibromatose plexiforme foi estabelecido. A próxima etapa consistiu na eliminação das demais formas sindrômicas e não sindrômicas. O exame físico revelou ausência de manchas “café com leite”, de sardas inguinais e femorais. Além disso, todos os critérios diagnósticos para neurofibromatose tipo I foram eliminados e nenhum outro tumor cutâneo ou subcutâneo foi encontrado.

Uma tomografia computadorizada de corpo total foi feita e não mostrou tumor na glândula adrenal, tumores abdominais ou pélvicos. Fizeram‐se tomodensitometria computadorizada e ressonância magnética do osso temporal, que não mostraram vestígios de schwannomas ou outros tumores dos nervos cranianos.
A avaliação do prognóstico foi baseada no resultado cirúrgico e no risco de recorrência e transformação maligna. Nenhuma mudança foi feita na intervenção terapêutica após a confirmação diagnóstica. O resultado da avaliação da paralisia do nervo facial periférico foi estabelecido no pós‐operatório, através da eletromiografia.
O paciente mostrou grande tolerância ao comprometimento do nervo facial e aderiu às medidas de proteção oftalmológica. Nenhum evento adverso ou imprevisto foi observado.
DiscussãoOs tumores neurogênicos do sétimo nervo craniano são raros e a localização na parótida parece ser ainda mais rara. A incidência estimada varia de 0,2% a 1,5%.1–3 O neurofibroma plexiforme solitário é uma das formas mais raras: apenas três casos de neurofibroma foram citados na literatura em inglês.1
É um tumor que depende de uma camada nervosa.4 O diagnóstico pré‐operatório é difícil devido à variação na apresentação clínica e sua estreita relação com o local do nervo envolvido.3
Não há sintoma patognomônico associado ao neurofibroma plexiforme intraparotídeo. Dor e problemas motores faciais, como paralisia ou espasmos, estão associados na maior parte. A citologia positiva é escassa, já que as células são bem aderentes.1,4 Nenhum achado de imagem é patognomônico de neurofibromas plexiformes.3 Os neuromas podem ter uma apresentação sindrômica, como parte da doença de Recklinghausen ou como múltiplos neurofibromas sem doença de Recklinghausen, ou como neurofibromas solitários.3,4
Além das manchas “café com leite”, outros tumores cutâneos e subcutâneos ou sinais neurológicos focais também estão presentes na maioria dos casos.5
O diagnóstico é estabelecido com base na história médica pessoal e familiar, bem como nos achados clínicos e radiológicos. Os neurofibromas sindrômicos são eliminados primeiro, seguidos pelas formas não sindrômicas.1,2,6 O principal tratamento dos neurofibromas plexiformes solitários consiste na sua excisão cirúrgica.1–6
Em escala macroscópica, os neurofibromas não encapsulados e múltiplos geralmente não incluem axônios. No entanto, os schwannomas são encapsulados, principalmente os solitários, aderidos aos nervos ou cercados por eles. Enquanto os neurônios apresentam sinais de degeneração, como necrose e alterações císticas. 1,4
Porém, histologicamente, os neurinomas e schwannomas apresentam algumas características particulares. Os neurofibromas têm estruturas muito mais frouxas do que os schwannomas (Sullivan et al.). Além disso, por um lado, os schwannomas apresentam‐se em dois padrões, Antoni A e Antoni B.3,5 No Antoni A são encontradas células fusiformes, com núcleos alongados dispostos em ondas, movimentos e espirais, que se unem em fitas trançadas. Na seção transversal, as células cilíndricas estão organizadas em um padrão semelhante a uma paliçada ao redor do citoplasma no centro, chamado de corpo de Verocay.3,5
No Antoni B há presença de tecido mais frouxo, sem qualquer arranjo em particular.3,5
Por outro lado, os neurofibromas são caracterizados por quase nenhuma estrutura organizada de células fusiformes em um tecido frouxo de matriz colagenosa, que contém fibras nervosas dentro do tumor.3,5 A vigilância básica é anual, desde que o tumor esteja estável.5 Quando o exame da lesão mostra anormalidades, uma ressonância magnética e/ou PET scan é indicada. Se forem encontrados sinais de malignidade, uma biópsia será indicada e a decisão terapêutica será baseada nos achados histológicos.5
Embora a questão da função do nervo facial seja crucial para o tratamento de neoplasias neurogênicas benignas do nervo facial, os tumores malignos raros precisam de remoção cirúrgica com margens cirúrgicas.
A presença de fraqueza pré‐operatória do nervo facial é um fator importante no prognóstico pós‐operatório.1,3 Entretanto, quando a função do nervo facial está intacta, surgem controvérsias sobre o manejo. Alguns autores recomendam a abstenção quando todos os parâmetros indicam a natureza benigna do tumor, caso em que protocolos de seguimento para tumores neurogênicos benignos parecem ser uma opção discutível, através de eletroneurografia e imagens radiológicas.3,5
Sempre que a função do nervo é prejudicada, suspeita‐se de malignidade e investigações adicionais devem ser feitas.3,5 O risco de transformação maligna no neurofibroma plexiforme solitário é menor do que nas formas sindrômicas.1,3,4 A maior parte das transformações sarcomatosas ocorrem em tumores associados a NF1 profundamente enraizados.1,6
ConclusãoO neuroma plexiforme solitário é uma forma muito rara de tumor da porção parotídea do nervo facial. Considerando as repercussões funcionais do tratamento cirúrgico, os médicos devem estar cientes de sua presença para preparar um plano terapêutico e de acompanhamento abrangente.
Conflitos de interesseOs autores declaram não haver conflitos de interesse.
Como citar este artigo: Hmidi M, Cherrabi K, Sinaa M, Nadour K. The challenging management of an assident parotid tumor: a case of solitary plexiform neurofibroma of the parotid facial nerve. Braz J Otorhinolaryngol. 2022;88:484–6.
A revisão por pares é da responsabilidade da Associação Brasileira de Otorrinolaringologia e Cirurgia Cérvico‐Facial.
gology tem o prazer em homenagear os revisores


