
Introdução: A síndrome de Turner (ST) é causada por uma deleção total ou parcial de um cromossomo X, ocorrendo em 1:2.000 até 1:5.000 meninas nascidas vivas. A perda auditiva é uma de suas principais manifestações clinicas. Entretanto, existem poucos estudos na literatura descrevendo essa associação.
Objetivo: Rever o conhecimento atual sobre a epidemiologia, etiologia, manifestações clínicas e diagnósticas da deficiência auditiva em pacientes com ST.
Métodos: A pesquisa bibliográfica, realizada entre 1980-2012, utilizou os bancos de dados Medlinee Lilacs identificando os principais artigos que relataram associação entre ST e deficit auditivo e suas repercussões clínicas.
Conclusões: Otite média de repetição, disfunção das trompas de Eustáquio, perdas auditivas condutivas durante a infância e perdas auditivas sensorioneurais a partir da adolescência são os distúrbios audiológicos mais comuns na ST. O cariótipo parece ter relação com a perda auditiva, com estudos mostrando aumento da prevalência de perda auditiva em pacientes com monossomia 45,X e isocromossomos 46,i(Xq). Estudos morfológicos da cóclea são necessários para ajudar a esclarecer a etiologia da perda auditiva sensorioneural.
Introduction: Turner's syndrome (TS) is caused by a partial or total deletion of an X chromosome, occurring in 1:2,000 to 1:5,000 live born females. Hearing loss is one of its major clinical manifestations. However, there are few studies investigating this problem.
Objectives: To review the current knowledge regarding the epidemiology, etiology, clinical manifestations and diagnosis of hearing impairment in patients with TS.
Methods: A bibliographic search was performed in the Medline and Lilacs databanks (1980-2012) to identify the main papers associating Turner's syndrome, hearing impairment and its clinical outcomes.
Conclusions: Recurrent otitis media, dysfunction of the Eustachian tube, conductive hearing loss during infancy and sensorineural hearing loss in adolescence are the audiologic disorders more common in ST. The karyotype appears to be important in the hearing loss, with studies demonstrating an increased prevalence in patients with monosomy 45,X or isochromosome 46,i(Xq). Morphologic studies of the cochlea are necessary to help out in the clarifying the etiology of the sensorineural hearing loss.
Introdução
A síndrome de Turner (ST) é uma doença rara, com variabilidade genotípica e fenotípica conhecida1 e que afeta de 1:5000 a 1:2000 meninas nascidas vivas.2-6 Esta síndrome é causada por uma deleção parcial ou total de um cromossomo X. Cerca de metade dos indivíduos afetados tem cariótipo 45,X, 20-30% exibem mosaicismo e o restante sofre anormalidades estruturais.7-9
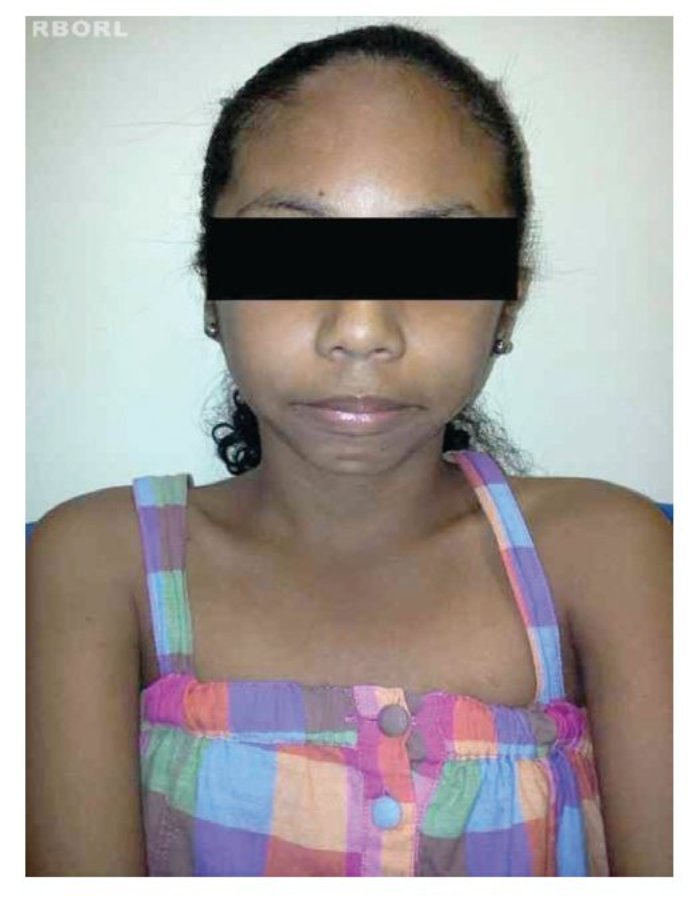
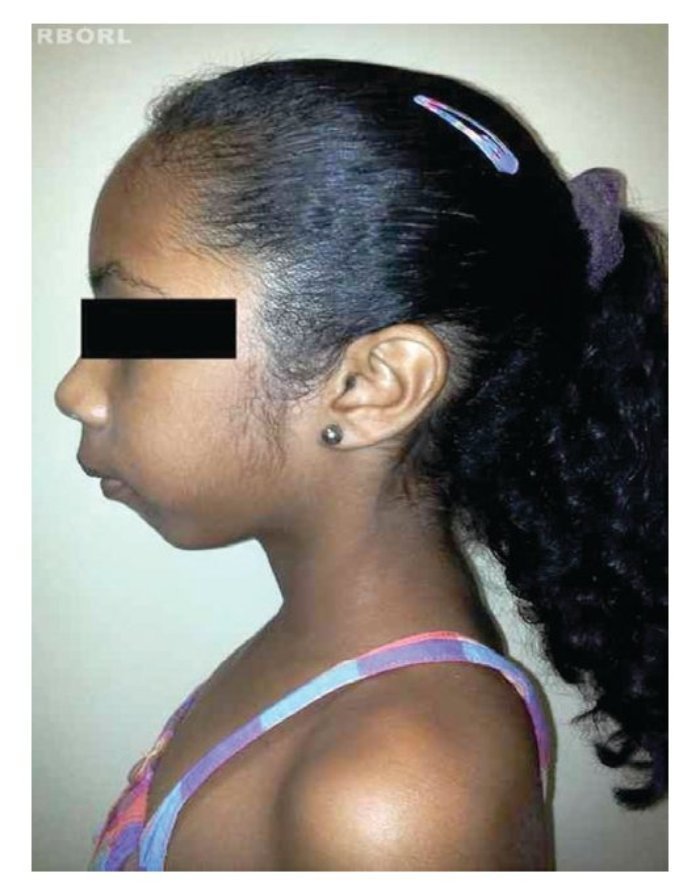
A perda completa ou parcial do cromossomo X provoca as manifestações fenotípicas da síndrome de Turner,8 incluindo baixa estatura, pescoço alado, rebaixamento da linha posterior de implantação dos cabelos, cubitus valgus, malformações cardíacas (p.ex., coartação da aorta, dilatação da raiz aórtica), malformações renais (p.ex., rim em ferradura) e disgenesia ovariana2,4,10,11(figs. 1 e 2).
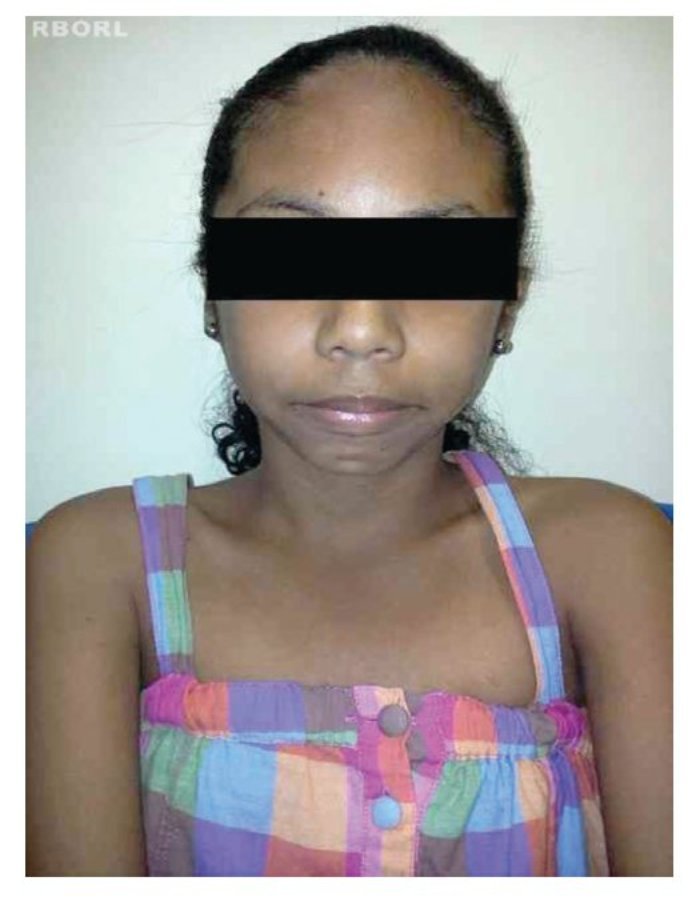
Figura 1 Vista frontal de paciente com syndrome de Turner.
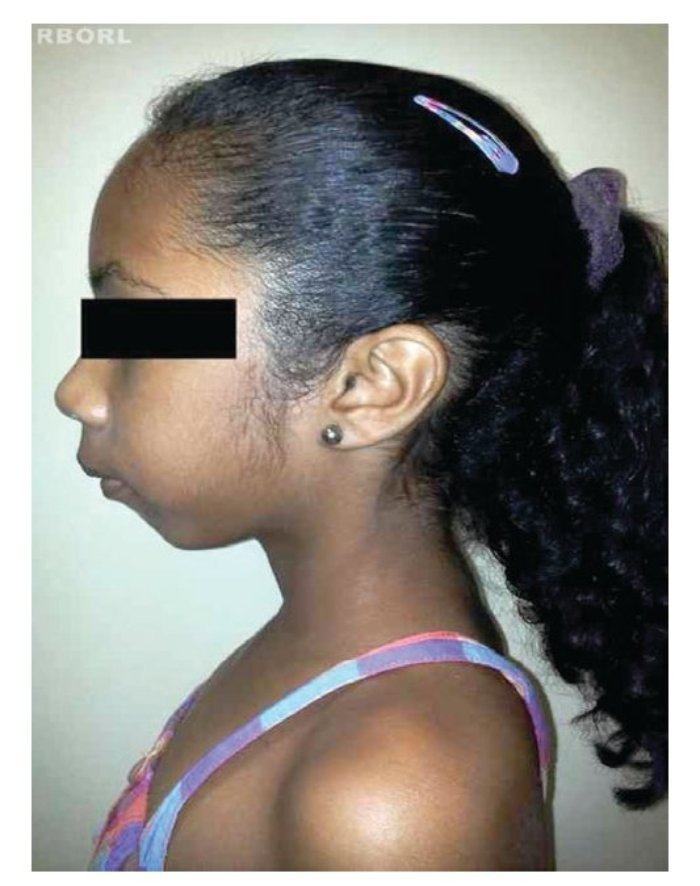
Figura 2 Vista lateral de paciente com syndrome de Turner.
A associação entre otite média, perda da audição e ST foi descoberta no início dos anos 60, sendo confirmada por estudos subsequentes.12 Sabe-se que indivíduos com ST apresentam incidência mais elevada de doenças da orelha média e problemas auditivos, em comparação com indivíduos não portadores dessa síndrome.13 O comprometimento auditivo associado foi descrito nas formas condutiva e sensorioneural, sugerindo envolvimento tanto da orelha média como da interna.13
Recentemente, alguns estudos descreveram o papel do gene SHOX (gene contendo homeobox para baixa estatura) na fisiopatologia da baixa estatura e dos problemas auditivos em pacientes com ST.14 A baixa estatura decorre principalmente da haploinsuficiência do SHOX, caracterizando-se por uma defasagem de aproximadamente 20 cm em relação à previsão da altura adulta.15
O gene SHOX pertence a uma família de genes homeobox, reguladores transcricionais e controladores-chave do processo do desenvolvimento. Esse gene se expressa no interior da faringe, envolvendo o primeiro e segundo arcos embrionários, a partir da sexta semana de gestação. Esses arcos se desenvolvem formando a maxila, a mandíbula e os ossículos da orelha média; os músculos envolvidos na abertura da trompa de Eustáquio, no amortecimento dos sons, na mastigação, na modulação da tensão do palato mole e na mudança das expressões faciais; e formam ainda a maior parte da língua e da orelha externa.14 Portanto, em pacientes com ST, a haploinsuficiência da expressão de SHOX é uma possível explicação de achados como palato ogival, orelhas salientes, otite média crônica, apneia obstrutiva do sono, aumento da sensibilidade aos ruídos e problemas como aprender a sugar, soprar, comer e proferir sons articulados.7,14
Otite média crônica grave é ocorrência comum em pacientes com ST, e acredita-se que isso decorra da obstrução da drenagem, seja por insuficiência linfática ou por displasia esquelética. A otite pode estar associada a uma perda auditiva condutiva na infância, que geralmente desaparece à medida que a frequência de otites diminue no estágio adulto jovem. Entretanto, a perda auditiva sensorioneural afeta muitos adultos jovens com ST e piora com o passar do tempo. Assim, uma triagem audiológica e cuidados específicos são importantes, tanto em meninas como em mulheres com ST.15
Dos muitos problemas crônicos para a saúde que devem ser enfrentados por pessoas com ST, as doenças otológicas e a perda auditiva estão entre os mais significativos e complexos. Embora vários estudos transversais, sobretudo retrospectivos, tenham descrito uma crescente prevalência de otite média e de perda auditiva em meninas em idade escolar e em mulheres com ST, não foi ainda publicado qualquer estudo prospectivo em grande escala que investigasse a prevalência e a história natural das doenças otológicas e da perda auditiva em bebês e na infância.16
Nesse artigo, objetivou-se revisar a literatura médica no que tange à epidemiologia, etiologia, manifestações clínicas e diagnóstico do comprometimento auditivo em pacientes com ST.
Nesse artigo, objetivou-se revisar a literatura médica no que tange à epidemiologia, etiologia, manifestações clínicas e diagnóstico do comprometimento auditivo em pacientes com ST.
Métodos
Foram pesquisados as bases de dados Medline, Lilacs, ProQuest, Cochrane e Embase em busca de estudos publicados entre 1980 a 2012. Foram utilizadas as palavraschave a seguir, combinadas por diferentes maneiras: síndrome de Turner; perda auditiva; disfunção auditiva; comprometimento auditivo; otite média; e avaliação audiológica. A revisão da literatura incluiu consensos, editoriais, artigos originais e artigos de revisão escritos em inglês ou português. A princípio, os estudos foram selecionados com base em seus títulos e resumos. Os desfechos desejados foram perda auditiva, disfunção auditiva e suas manifestações clínicas. Foi realizada uma revisão das referências bibliográficas de todos os artigos encontrados, com a finalidade de expandir a pesquisa. Sessenta artigos atenderam aos critérios de inclusão e, assim, foram pesquisados e incluídos na presente revisão da literatura.
Perda auditiva na síndrome de Turner
A otite média é extremamente comum em meninas com ST, e cerca de 15% das mulheres adultas com ST sofrem perda auditiva.17 A perda auditiva na síndrome de Turner pode ser do tipo condutivo, sensorioneural ou misto. Pelo menos 25% dos adultos sofrem perda auditiva, necessitando de aparelho de amplificação sonora. A otite média recorrente é comum, e supõe-se que esta ocorra como resultado de uma estrutura/funcionamento anormal da trompa de Eustáquio. Infecções recorrentes acarretam a formação de tecido cicatricial no tímpano e perda auditiva condutiva, podendo ocorrer mesmo na primeira infância. À medida que vão crescendo, as pacientes se tornam suscetíveis a uma perda auditiva sensorioneural progressiva, que pode estar relacionada ao envelhecimento prematuro. Considerando que essas perdas podem ser sutis, mas funcionalmente comprometedoras tais pacientes deverão passar por avaliações rotineiras da audição até a vida adulta, particularmente no caso de mau desempenho escolar.18
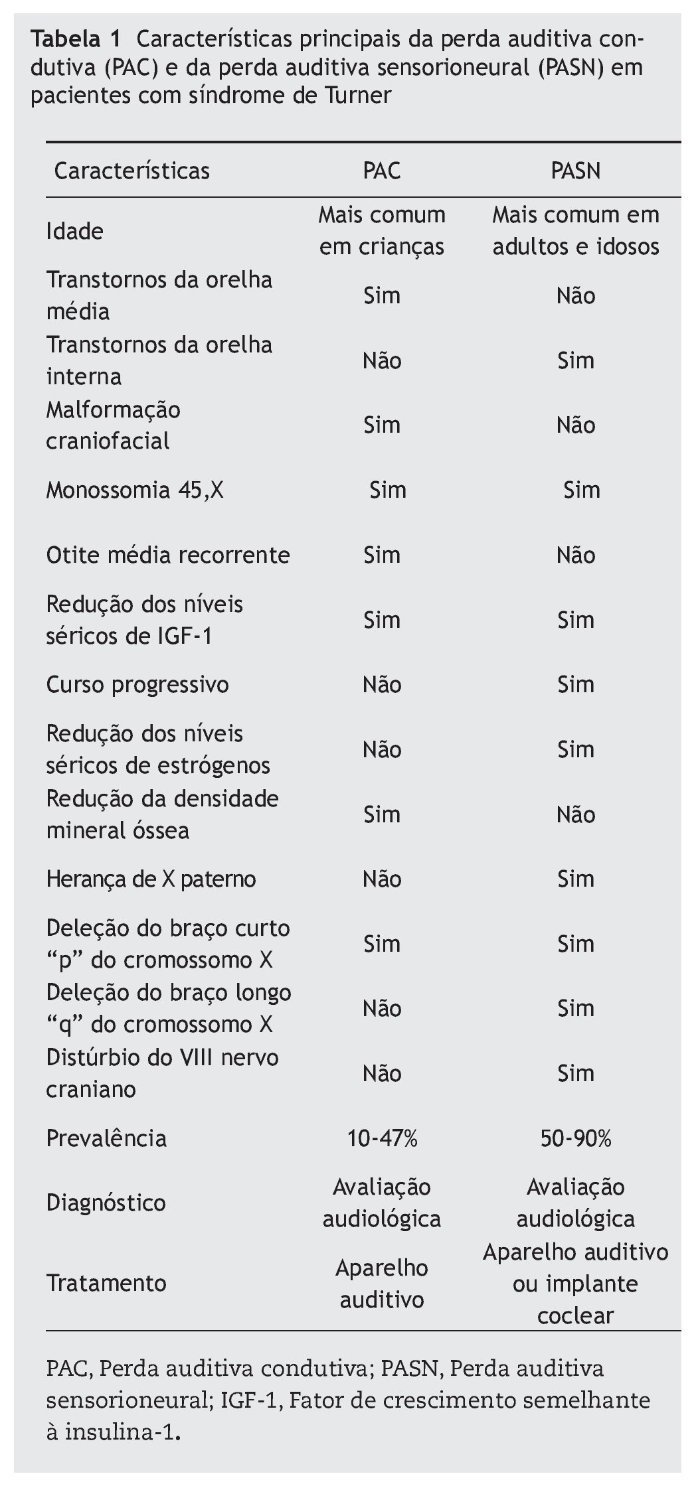
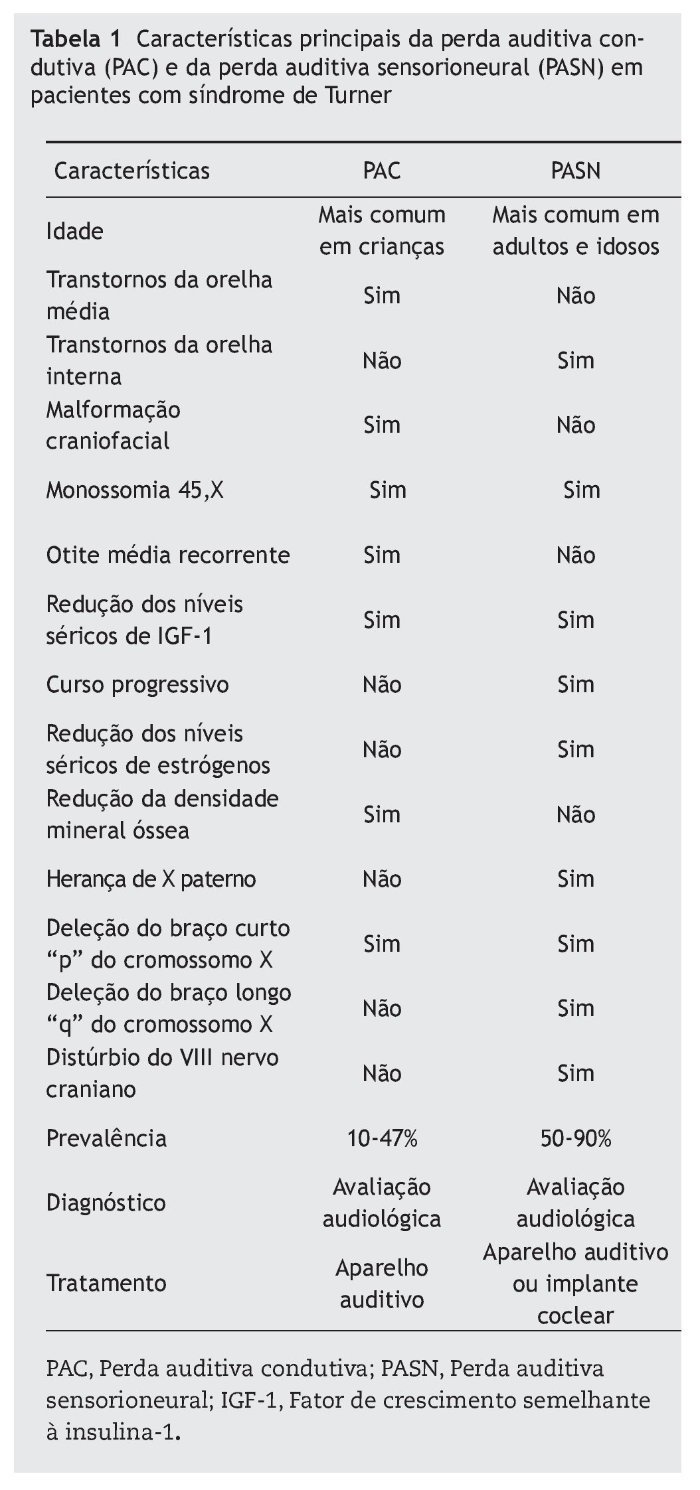
A tabela 1 resume as principais características da perda auditiva condutiva e sensorioneural na síndrome de Turner.
Perda auditiva condutiva
Epidemiologia
Estima-se que a incidência de perda auditiva condutiva (PAC) em pacientes com ST possa chegar a 80%, e que a disfunção da trompa de Eustáquio e otite média afetem até 88% dos pacientes.19,20 A elevada prevalência de otite média em pacientes com ST é um fator importante que possivelmente possa influenciar o fenótipo neuromotor individual levando a transtornos do equilíbrio em crianças que, afora isso, seriam consideradas saudáveis.21
Além disso, no contexto envolvendo PAC em portadores de ST, surdez é ocorrência comum e subnotificada.22
A PAC é mais prevalente em crianças e adolescentes, em comparação com adultos, e a frequência de infecções otológicas diminui com a idade e com o crescimento das estruturas faciais.23 Um melhor conhecimento dos problemas auditivos no cenário da ST e o adequado tratamento otológico têm contribuído para um recente decréscimo na PAC entre as complicações de doenças da orelha média.24
Etiologia
A principal razão para a ocorrência de PAC em portadores de ST é uma otite média persistente e recorrente acompanhada por efusão, infecção crônica da orelha média e degeneração ossicular, seguida por uma otite média crônica e colesteatoma na orelha média.25 Essas condições são resultantes da disfunção da trompa de Eustáquio e da malformação anatômica da base do crânio, causando uma drenagem deficiente e ventilação inadequada da orelha média, além de facilitar a invasão da orelha média por microrganismos nasofaríngeos.10,25,26 Nesses pacientes, a disfunção palatina pode ser exacerbada pela remoção das tonsilas faríngeas.5
Anormalidades da orelha externa - aurículas com baixa implantação e/ou em cúpula, inclinação inferior anormal da hélice, estenose anterógrada e inclinação superior dos condutos auditivos externos - são as anomalias auditivas mais comuns observadas em pacientes portadores de ST.6,27 Além disso, a síndrome de Turner também está associada a uma anomalia do estribo, e acredita-se que pacientes com ST sofram maior incidência de fenda palatina.28 Embora os estudos de tomografia computadorizada da orelha média não tenham revelado anomalias estruturais,29 Bergamaschi et al.30 verificaram uma associação significativa entre PAC e anormalidades craniofaciais, especialmente pescoço alado, micrognatismo, palato ogival e orelhas com baixa implantação.
Portanto, em pacientes com ST, os transtornos da orelha média são secundários a anormalidades na anatomia da orelha, como: (1) hipoplasia linfática, causando efusão linfática persistente na orelha média, o que compromete a aeração e a drenagem; (2) óstio timpânico da trompa anormal; (3) hipotonia do músculo tensor do véu palatino; e (4) braquicefalia e palato ogival, causando um posicionamento anormalmente horizontal das trompas de Eustáquio e possível disfunção palatina.20,31,32
Afora a inflamação da orelha média, pesquisa recentemente publicada demonstrou que a perda auditiva, as infecções da orelha média e as malformações da orelha externa em pacientes com ST estão relacionadas ao grau de deleção do braço "p" (braço curto do cromossomo X), sugerindo que a perda auditiva também esteja provavelmente associada ao cariótipo.26 Alguns estudos sugeriram que a monossomia 45,X e o isocromossomo 46,i(Xq) estão significativamente associados à ocorrência de PAC em pacientes com ST,10 indicando um possível envolvimento da perda de um gene no braço curto (p) do cromossomo X, por exemplo, SHOX.33 Mas, outros autores não encontraram uma associação entre PAC e cariótipo.30
Os problemas otológicos que levam à PAC também estão sendo associados a um decréscimo nos níveis séricos do fator de crescimento semelhante à insulina-1 (IGF-1). Barrenäs et al.34 observaram que baixos níveis de IGF-1 tinham correlação com uma elevada ocorrência de otite média. A complementação com hormônio do crescimento foi efetiva em termos de melhora da otite média, sugerindo que esse problema poderia estar sendo causado pela hipoplasia da base craniana e da cavidade mastoidea.35 Além disso, Davenport et al.16 encontraram evidências de que o tratamento com GH não aumenta a ocorrência de problemas otológicos em meninas jovens com ST. Infelizmente, estudos acurados tratando do efeito do uso do hormônio do crescimento e de desfechos auditivos serão praticamente impossíveis, pois a maioria dos pacientes com ST recebe complementação de hormônio do crescimento.19
Ao que parece, nestes pacientes a etiologia da otite média recorrente não está relacionada a uma disfunção imunológica.34
Manifestações clínicas
Aproximadamente 30% das meninas com dez anos ou menos sofrem de perda auditiva condutiva,36 mas à medida que as infecções da orelha média vão diminuindo com a idade, ao final da adolescência essa complicação terá praticamente desaparecido.37 Ao contrário da perda auditiva sensorioneural, a PAC não progride com o passar do tempo.
Além dos achados citados acima, a incidência de colesteatoma é mais elevada em casos de ST do que na população em geral, sendo bilateral em 90% dos casos e diagnosticada, na média, por volta dos 15,5 anos.30 Mulheres com ST e com baixa densidade mineral óssea (DMO) e comprometimento auditivo, particularmente do tipo condutivo, estão em maior risco de sofrer fraturas ósseas. A melhora na DMO e na capacidade auditiva pode ajudar a reduzir o risco de fratura.38
Perda auditiva sensorioneural
Epidemiologia
A doença auditiva em pacientes adultos com ST complica-se ainda mais com uma perda auditiva sensorioneural (PASN) progressiva.1 Geralmente, a PASN tem início no final da adolescência e início da vida adulta, e progride gradualmente,10 sendo uma característica marcante da síndrome de Turner.22 Na literatura, as incidências variam de 11-67% de perda auditiva sensorioneural em pacientes com até 42 anos de idade1. Apesar disso, em um estudo, 90% dos pacientes adultos (44%) com síndrome de Turner sofriam de perda auditiva sensorioneural. A perda foi considerada clinicamente significativa em dois terços dos indivíduos, e 27% deles necessitaram da ajuda de um aparelho auditivo.5 Por outro lado, Kavoussi et al.39 observaram que a perda auditiva sensorioneural é extremamente comum em mulheres com ST, afetando até 90% dos indivíduos e, consequentemente, causando dificuldades nas tarefas escolares e nas interações sociais da adolescência.
A incidência de PASN varia de 16-60% para perdas nas frequências médias e 11-40% para perdas nas frequências altas.20 Embora o padrão audiométrico clássico da PASN em pacientes com ST seja a perda auditiva nas frequências médias, alguns autores relatam incidências mais elevadas de padrão de perda nas frequências altas.26 A discrepância entre esses dados pode estar relacionada à diferença na definição do padrão de rebaixamento. Embora investigadores, como Hultcrantz et al.40, tenham definido o padrão como uma elevação do limiar excedendo 5 dB nas frequências médias (0,5-2 kHz), outros, como Morimoto et al.,26 o definem como um limiar excedendo 15 dB. Por outro lado, outros pesquisadores chegam mesmo a um limiar excedendo os 20 dB.25,26 Outra explicação possível para variação dos resultados é que a perda progressiva auditiva nas frequências altas oculta a queda audiométrica nas frequências médias.26 Finalmente, alguns estudos podem ter incluído crianças demasiadamente jovens, nas quais o padrão de queda audiométrica talvez ainda não tenha se desenvolvido.25
Etiologia
As causas da perda auditiva nas frequências altas em pacientes com ST permanecem um motivo de controvérsia. Os mecanismos mais frequentemente estudados são: idade, cariótipo (p.ex., monossomia vs. dissomia, origem paterna do cromossomo X, supressão de Xp), deficiência de estrogênio e/ou hormônio do crescimento e deficiência de IGF-1 e anormalidades cocleares.10,26,37
A PASN foi atribuída a um efeito de dose ligado a X, porque o declínio auditivo é mais comum em casos de ST com monossomia completa para o braço curto (braço "p"), por exemplo, cariótipos 45,X e 46, Xi(Xq), em comparação com indivíduos com deleções menores de X.41,42 É possível que genes como SHOX possam induzir um ciclo celular mais tardio, com menor número de células sensitivas na cóclea por ocasião do nascimento, o que resultaria em disfunção coclear.26
Na maioria dos casos, pacientes com ST herdam seu cromossomo X intacto (Xintacto) de suas mães.43 O imprinting genômico, um fenômeno pelo qual existe uma expressão diferencial de alguns genes, dependendo de sua origem cromossômica, foi demonstrado em pacientes com ST.44 Seguindo esse raciocínio, Hamelin et al.41 relatam que, depois de um teste simples de audiometria, a prevalência de PASN foi maior em pacientes com ST que herdaram o cromossomo X paterno (Xpaterno), em comparação com indivíduos Xmaterno, demonstrando evidência para um efeito ligado a X na ocorrência de PASN. Ademais, indivíduos com ST com um cariótipo 46,X,i(Xq) exibem a mais alta prevalência de PASN, provavelmente porque esses indivíduos herdam mais frequentemente o cromossomo Xpaterno.41,44 Esses achados sugerem que os genes expressos no cromossomo Xmaterno podem proteger contra PASN.41
Um PEATE (Potencial Evocado Auditivo de Tronco Encefálico) anormal em um paciente com ST sugere comprometimento da função coclear, que pode ter sido causado por alguma perturbação neuropática, ou por defeitos do gânglio coclear.13,25 De fato, "camundongos Turner" exibem perda da resposta auditiva no tronco cerebral com o passar do tempo.40
Especulou-se que os estrógenos tenham efeito protetor da audição.45 Wang et al.46 observaram que um camundongo knockout para o receptor-β de estrogênio (BERKO) desenvolve hipocelularidade neural no córtex somatossensitivo, e Meltser et al.47 relataram que o camundongo BERKO é mais suscetível a problemas auditivos em seguida a problemas acústicos. Coleman et al.48 observaram melhora do PEATE em ratas ovariectomizadas em seguida à reposição de estrogênio. Ostberg et al.19 relataram que o risco relativo de ocorrência de perda auditiva sensorioneural em pacientes com síndrome de Turner foi mais alto para os casos em que a reposição de estrogênio teve início depois dos 13 anos de idade, ou em mulheres com deficiência de estrogênio por mais de dois anos.
No entanto, não se sabe se o comprometimento auditivo associado à deficiência de estrogênio se deve à mineralização deficiente da cápsula coclear, ou se decorre da falta de estimulação dos receptores estrogênicos, resultando em um desenvolvimento anormal da orelha interna.38 Outros investigadores que estudaram pacientes com ST não puderam confirmar esse efeito protetor do estrogênio na audição.19,30
Manifestações clínicas
Comumente, a PASN nas frequências médias em pacientes com ST se inicia na segunda ou terceira década (final da adolescência e início da vida adulta), embora já tenha havido relatos de casos em crianças com menos de dez anos.20
Com a passagem dos anos, a PASN evolui para uma perda auditiva nas frequências altas, resultando, por volta dos 40 anos, em uma audição comparável à de mulheres de 60 anos, não portadoras de ST.32,41 Ao avaliar pacientes com ST, é importante que sejam excluídas as causas comuns de perda auditiva nas frequência mais altas, como a ototoxicidade e a exposição a sons intensos.
Perda auditiva mista
A perda auditiva mista (PAM) em pacientes com ST se caracteriza pela associação de transtornos da orelha média e PASN. PAM é diagnosticada quando os limiares da condução óssea são superiores a 15 dB HL e os limiares da condução aérea são superiores a 25 dB, com uma diferença aérea-óssea (gap) igual ou superior a 15 em pelo menos uma frequência.49 A literatura sobre ST se torna ainda mais confusa pela inclusão das perdas auditivas mistas, não diferenciadas em seus componentes condutivo e sensorioneural, e frequentemente sem qualquer outra identificação da contribuição da otite média crônica para os níveis rebaixados da condução óssea.20
Triagem para perda auditiva na síndrome de Turner
Tendo em vista que a doença otológica causa profundo impacto na qualidade de vida em pacientes com ST,16 os pais e o paciente (por ocasião da adolescência) devem tomar ciência da alta prevalência de problemas auditivos quando do diagnóstico de ST.50 Além disso, o audiologista deve suspeitar de ST ao avaliar uma criança com PASN nas frequências médias ou com otite média crônica, especialmente se houver uma associação com baixa estatura, atraso na puberdade ou estigmas físicos.25,50
A elevada incidência de otite média aguda recorrente em meninas com ST enfatiza a importância de exames otológicos periódicos, não apenas para confirmar o diagnóstico de ST, mas também para monitorá-los para possível perda auditiva condutiva ou sensorioneural.8
A abordagem a seguir é sugerida para a triagem de transtornos otológicos em pacientes com ST: triagem neonatal com PEA-TC e primeira audiometria entre os 18-24 meses, seguida por audiometrias anuais e avaliações otorrinolaringológicas. Os pacientes com problemas otológicos devem ser encaminhados para acompanhamento especializado.23,30,31
É importante identificar a perda auditiva inicial em frequências acima de 8 kHz, antes que ela evolua para frequências críticas para a comunicação verbal. Por ocasião de um diagnóstico de deficiência auditiva por método convencional, é possível que já tenha ocorrido o comprometimento da comunicação.31 Assim, deve-se solicitar um teste audio-lógico que inclua frequências altas para todos os pacientes com ST com mais de três anos de idade, pois esse exame poderá diagnosticar a perda auditiva antes que esta se torne evidente em uma audiometria de rotina.25,31 Tendo em vista que a perda auditiva de frequência média é de, em torno, 18,8 dB - um grau de perda auditiva geralmente não detectado pela triagem auditiva básica de rotina de 20 dB - é importante obter limiares de condução aérea/óssea para toda criança com suspeita de ST.25
A impedânciometria é indicada para verificar as condições da orelha média (p.ex., a continuidade da membrana timpânica, a pressão na orelha média, presença de reflexo do músculo estapédio); a tomografia computadorizada do osso temporal é indicada em pacientes com doença progressiva da orelha média e colesteatoma; PEATE deve ser requisitado quando houver necessidade de investigar lesão coclear;30 e pode haver indicação de um exame de EOPD (Emissão Otoacústica por Produto de Distorção) para avaliar a função da microfonia coclear e verificar se as células auditivas externas estão funcionando corretamente.
Conclusões
Doença otológica e perda auditiva são frequentes em pacientes com ST. Os transtornos da orelha média se iniciam precocemente na infância, e a perda auditiva se manifesta na primeira década. Ambos podem evoluir para o comprometimento auditivo e para a incapacitação social (p.ex., deficiência da fala ou mesmo do desenvolvimento intelectual), caso não sejam precocemente diagnosticados e tratados. A única intervenção bem-sucedida para a redução da perda auditiva em pacientes com ST é um tratamento diligente e apropriado de seus problemas auditivos. Para o devido esclarecimento da etiologia da PASN, são necessários estudos morfológicos do órgão de Corti e do nervo vestibulococlear tanto na ST fetal como na adulta.
Conflitos de interesse
Os autores declaram não haver conflitos de interesse.
Recebido em 2 de novembro de 2012;
aceito em 23 de agosto de 2013
DOI se refere ao artigo: http://dx.doi.org/10.1016/j.bjorl.2013.08.002
¡î Como citar este artigo: Alves C, Oliveira CS. Hearing loss among with Turner's syndrome: literature review. Braz J Otorhinolaryngol.2014;80:257-63.
*Autor para correspondência.
E-mail: cresio.alves@uol.com.br (C. Alves).


