
O sarcoma de Ewing foi identificado por James Ewing, em 1921, como um tumor endotelial perivascular.1 Desde a década de 1980, acredita‐se que esse tumor se origine de células mesenquimais, mieloides ou multipotentes primitivas.2 Embora a maior parte se origine a partir dos ossos dos membros inferiores, também pode surgir em tecidos moles, tais como a região paravertebral. Esses pequenos tumores de células redondas foram denominados de sarcoma de Ewing (SE)/tumor neuroectodérmico primitivo periférico (PNET) e incluem SE de osso, SE extraesquelético, PNET e tumor de Askin.3
SE extraesquelético raramente ocorre na região da cabeça e do pescoço e responde por apenas 1 a 4% dos casos de SE. Geralmente afeta a mandíbula,4 mas há relatos na literatura de SE em seios maxilares.4,5 A distribuição entre os sexos é igual e quase metade dos pacientes tem entre 10 e 20 anos, enquanto 70% têm menos de 20.6 Os pacientes geralmente apresentam‐se com edema indolor e sintomas como anemia, leucocitose, perda de peso, congestão nasal (com base na localização), perda de visão e dor de cabeça.4 Aqui, descrevemos uma paciente do sexo feminino de 11 anos, diagnosticada com SE no seio maxilar. As características do tumor e a abordagem clínica são revistos na literatura.
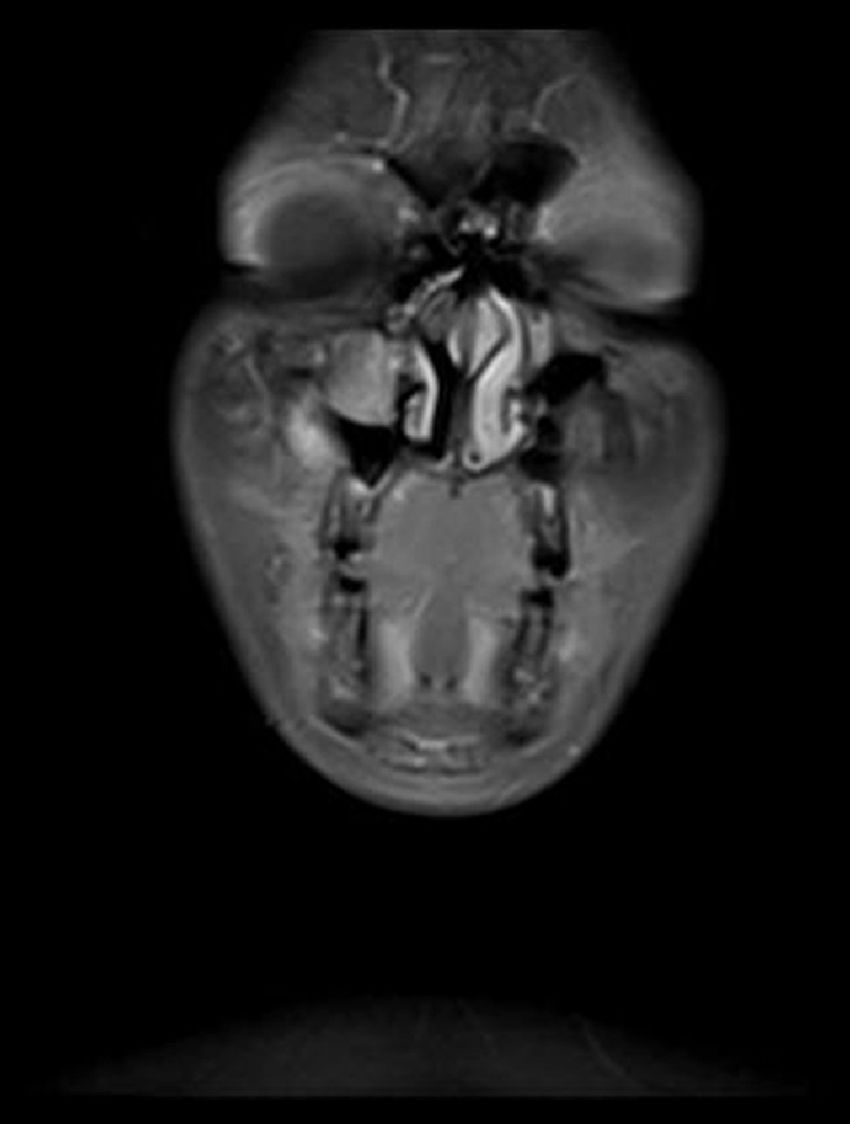
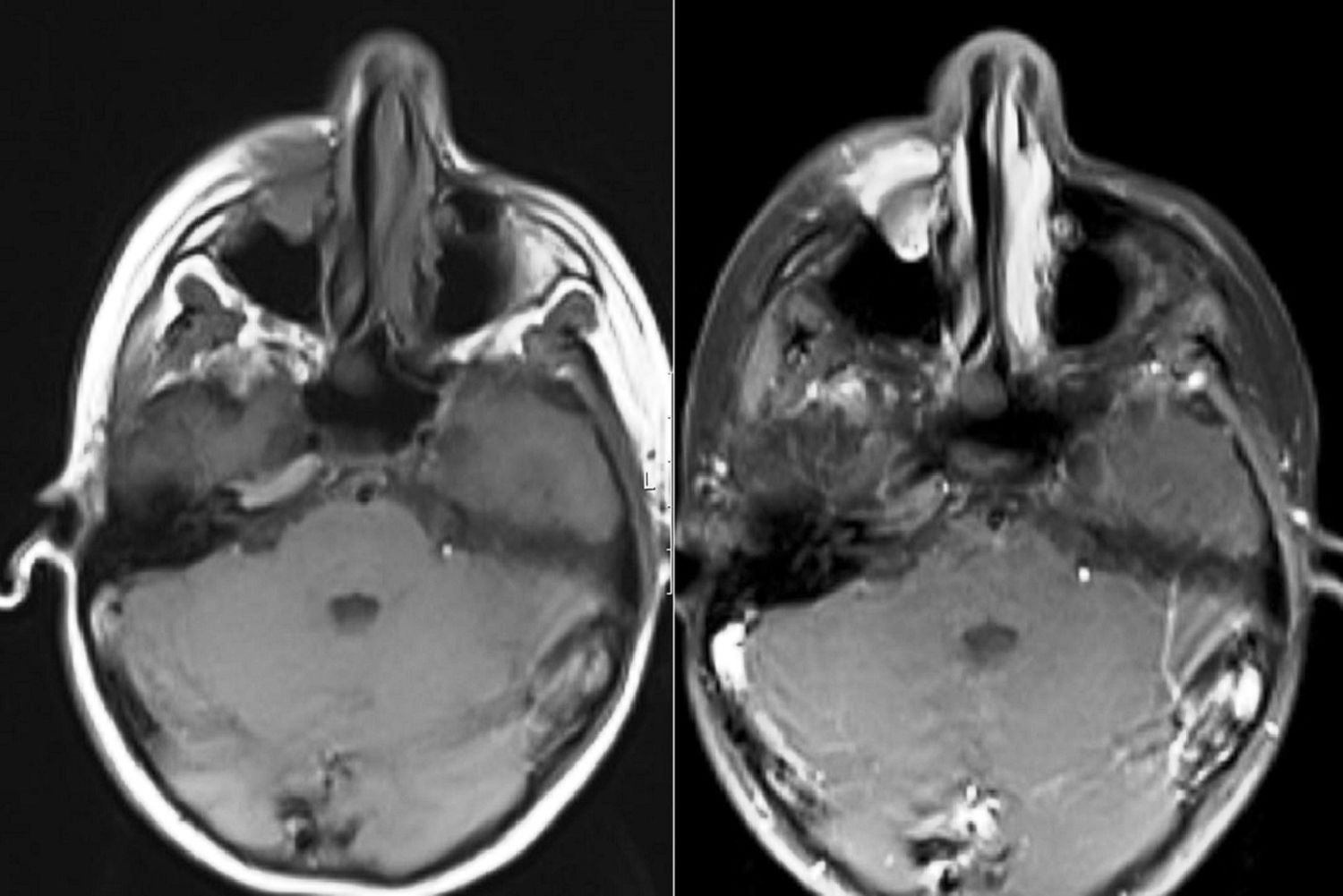
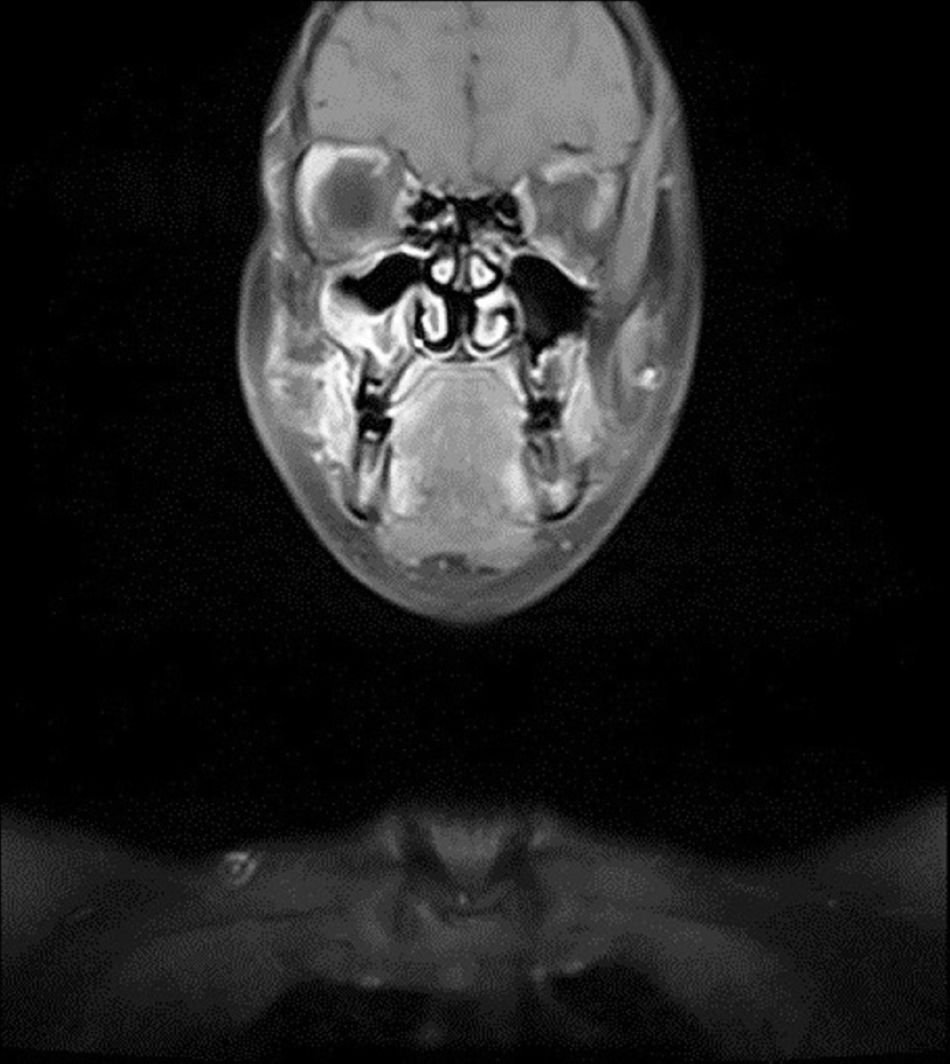
Relato de casoPaciente do sexo feminino, 11 anos, apresentou‐se à nossa clínica com queixa de edema na face direita. A história revelou que o edema havia surgido duas semanas antes e aumentara cada vez mais desde então. No exame físico, não houve outros resultados significativos, com exceção de uma lesão tumoral na face direita, imóvel, dura à palpação, que media 2×2cm. Os exames laboratoriais não mostraram alterações. Imagem por ressonância magnética (MRI) revelou uma lesão tumoral centralizada no seio maxilar direito, que se estendia até a parede anterior e os tecidos moles (figs. 1 e 2). Não foram identificadas metástases.


Devido à suspeita de malignidade, a paciente foi submetida a cirurgia aberta, com abordagem por meio de incisão sublabial, e as amostras da biópsia foram posteriormente submetidas a exame anatomopatológico.
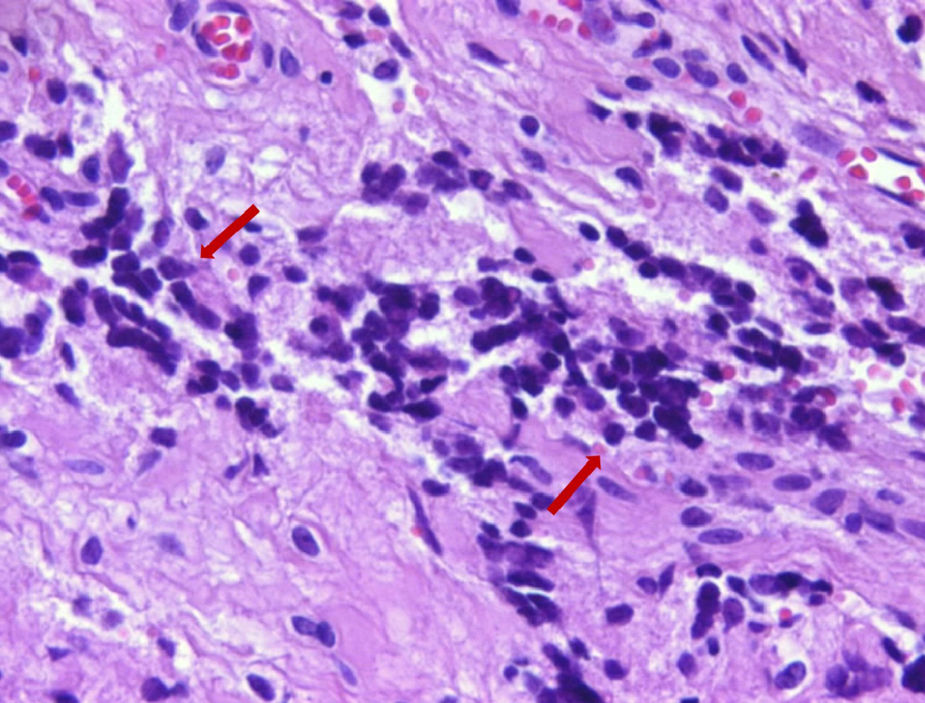
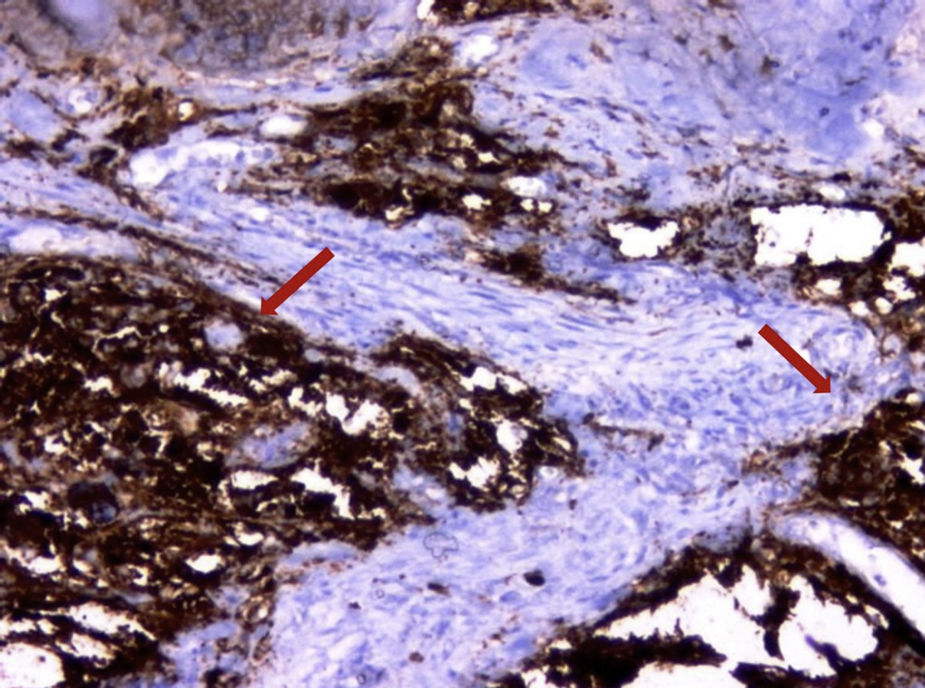
No exame histopatológico, foi constatada presença de pequenas células redondas compactas e uniformes, com alta proporção nuclear/citoplasmática e cromatina finamente dispersa, sem nucléolo proeminente, e numerosas figuras mitóticas. Áreas de necrose estavam presentes. No exame imuno‐histoquímico, as células tumorais foram fortemente positivas com CD99 e também com vimentina. Agrupamentos ocasionais apresentavam algumas células positivas para sinaptofisina e CD56. As células tumorais foram negativas com CD3, CD20, LCA, PanCK, miogenina, myoD1 e desmina. O índice proliferativo Ki‐67 foi de 30 e 50% em algumas áreas. Posteriormente, SE foi considerado o diagnóstico mais provável. Assim, o material da paciente foi submetido a patologia molecular para avaliação do clássico rearranjo de EWSR1‐FLI1. Os resultados da hibridização fluorescente in situ (FISH) para o rearranjo de EWSR1‐FLI1 confirmaram o diagnóstico de SE/PNET (figs. 3 e 4).
. (Hematoxilina & eosina,×400).")
 (200x). Legendas das figuras e setas corrigidas foram adicionadas às microfotografias.")
A paciente foi tratada com quimioterapia, a partir do protocolo de oncologia pediátrica do grupo para sarcoma de Ewing, com VDC/IE (V, vincristina 1,5mg/m2; D, doxorrubicina 75mg/m2;C, ciclofosfamida 1,2 gr/m2; I, Ifosfamida 1,8 gr/m2; E, etoposídeo 100mg/m2). Foram administrados 17 ciclos ao longo de 48 semanas. Durante o seguimento, após quatro meses, detectou‐se recorrência local. A paciente foi submetida a cirurgia aberta com o mesmo procedimento, com abordagem por meio de incisão sublabial. A ressecção foi estendida para alcançar com seguranças as margens tumorais negativas. A radioterapia foi adicionada ao tratamento após a cirurgia. A investigação de metástases foi feita por tomografia, com emissão de pósitrons (PET), e ressonância magnética, a cada seis meses. Além disso, a paciente foi acompanhada por exame clínico de rotina a cada três meses. Não houve constatação de recidiva ou metástase no 13° mês de seguimento (fig. 5).
Discussão
O SE extraesquelético é um tumor raro na região da cabeça e do pescoço. O diagnóstico diferencial inclui várias entidades que ocorrem nos ossos e tecidos moles do trato nasossinusal, como melanoma maligno, rabdomiossarcoma, carcinoma indiferenciado sinonasal, linfoma e neuroblastoma olfatório. Em nosso caso, considerando a idade da paciente, SE extraesquelético, rabdomiossarcoma e linfoma foram incluídos no diagnóstico diferencial. Rabdomiossarcoma e linfoma foram excluídos e o diagnóstico de SE extraesquelético foi confirmado na análise imuno‐histoquímica e Fish para rearranjo de EWSR1‐FLI1.7
A maioria dos SE tem t (11; 22) (q24; q12), o que corresponde a uma fusão entre o gene EWS (22q12) e o gene FLI1 (11q24), o qual é um fator de transcrição.
Além disso, quatro translocações diferentes são observadas: EWS‐ERG t (21;22) (q22;q12), EWS‐ETV1 t (7;22) (p22;q12), EWS‐E1AF t (17;22) (q12;q12) e EWSFEV t (2;22) (q33;q12), de acordo com a frequência, respectivamente.8,9 A detecção do rearranjo de ESWR1‐FLI1 não é diagnóstico para SE, uma vez que também é observado em rabdomiossarcoma.10 Neste caso, a avaliação histopatológica e imuno‐histoquímica, em conjunto com o teste molecular para rearranjo de EWSR1‐FLI1, proporcionou um diagnóstico definitivo. Ao avaliar um paciente no qual o diagnóstico de SE é suspeitado ou aqueles com diagnóstico definitivo, métodos de imagem apropriados devem ser aplicados como prioridade. A tomografia computadorizada é usada para monitorar áreas de destruição óssea e propagação da massa, enquanto a RM é um método ideal para a avaliação do envolvimento do tecido mole ao redor da lesão primária.9 Em nossa paciente, a extensão do tumor foi avaliada por RM.
O objetivo do tratamento do SE é manter todas as funções normais, prevenir recorrências e sequelas em longo prazo e eliminar a doença. O tratamento recomendado é uma combinação de cirurgia com quimioterapia e radioterapia. O diagnóstico precoce seguido por ampla ressecção, quimioterapia e radioterapia pode deixar o paciente livre da doença por um longo tempo. Recentemente, tem sido relatado que o uso de terapia de feixe de prótons proporciona um controle local da doença e evita possíveis complicações da radioterapia sobre os tecidos adjacentes (tais como perda de visão e complicações intracranianas).9,10 Neste caso, depois da recorrência, adicionamos a radioterapia ao tratamento por quimioterapia após a reoperação.
ConclusãoO sarcoma de Ewing é de ocorrência rara no seio maxilar. Cirurgia, quimioterapia e radioterapia devem ser usadas em combinação para tratar essa lesão, que se origina de tumores de grau elevado. Os pacientes devem ser cuidadosamente acompanhados devido à possibilidade de recorrência local e metástases a distância.
Conflitos de interesseOs autores declaram não haver conflitos de interesse.
Como citar este artigo: Yaprak N, Toru HS, Ozbudak IH, Derin AT. Primary extraskeletal Ewing's sarcoma of the maxillary sinus. Braz J Otorhinolaryngol. 2019;85:538–41.
A revisão por pares é da responsabilidade da Associação Brasileira de Otorrinolaringologia e Cirurgia Cérvico‐Facial.
gology tem o prazer em homenagear os revisores


